
How to use CRC and Open Ondemand webportal to teach bioinformatics courses
Request an Allocation for a Course
The course instructor can fill out this form and attach a spreadsheet file containing the following information on each of the students from your class roster that will require access to the allocation's resources:
Name, Pitt Email (username@pitt.edu, no aliased emails)
The format of your course's user group name and slurm allocation will be SUBJECTNUMBER-YEAR(f/s) where the f/s denotes fall or spring semester. We will allocate 25K Sus and 5TB group shared storage to your course. Duration of computing time and storage: active for 1 term ((4 months). We will provide you the slurm allocation name and the storage location when we reply to your ticket. In the below demonstration, we will use course-2023s as the slurm allocation name and the storage location is /ix/genomics/demo. Note that there is an expiration date for the 5TB storage allocation. We will delete the 5TB storage allocation without discussing with the instructors or students.
If you are off-campus, the clusters are accessible securely from almost anywhere in the world via the PittNet Virtual Private Network (VPN), which is administered by Pitt IT. The VPN requires certain software to run on your system. We recommend Global Protect.
IMPORTANT NOTES: Pitt Wireless is off-campus by the definition of Pitt IT. VPN is required if you are using Pitt Wireless.
https://crc-pages.pitt.edu/user-manual/applications/application-environment/
Lmod will be used by cluster administrators to provide optimized builds of commonly used software. Applications are available to users through the Lmod modular environment commands. There are no default modules loaded when you log in. You can use this system to teach the course. You can also create conda environment and install your own tools.
The instructor can create a folder under the course storage to install conda environments.
[fangping@login0b ~]$ cd /ix/genomics/demo [fangping@login0b demo]$ mkdir software
We are using slurm as workload manager. To use the Sus from the course, you can use --account=course-2023s to charge resources used by this job to the specified course account. We recommend the instructor to provide slurm job templates.
Using R
https://crc-pages.pitt.edu/user-manual/applications/r%2Brstudio/
We have installed multiple R modules. You can use module spider r to view available modules and module spider r/version to show how to load the module. The latest r/4.3.0 can be loaded as:
[fangping@login0b demo]$ srun --account=course-2023s --pty bash [fangping@htc-1024-n3 demo]$ module load gcc/12.2.0 r/4.3.0 [fangping@htc-1024-n3 demo]$ R R version 4.3.0 (2023-04-21) -- "Already Tomorrow" Copyright (C) 2023 The R Foundation for Statistical Computing Platform: x86_64-pc-linux-gnu (64-bit) R is free software and comes with ABSOLUTELY NO WARRANTY. You are welcome to redistribute it under certain conditions. Type 'license()' or 'licence()' for distribution details. Natural language support but running in an English locale R is a collaborative project with many contributors. Type 'contributors()' for more information and 'citation()' on how to cite R or R packages in publications. Type 'demo()' for some demos, 'help()' for on-line help, or 'help.start()' for an HTML browser interface to help. Type 'q()' to quit R. >
Within each R module, various R packages and bioconductor packages have been installed. For the above r/4.3.0, the location of these packages is /ihome/crc/install/gcc-12.2.0/r/4.3.0/lib64/R/library. Within the R console, load the library to check whether it is already installed.
You can also install your own R packages. R searches the user’s path for libraries followed by the root installation. R will stop searching when it finds the first instance of the library within the path hierarchy.Use “.libPaths()” to check the searching path. For r/4.3.0, your local R packages will be installed under ~/R/x86_64-pc-linux-gnu-library/4.3. To allow all attendees to use the same R packages, we recommend that the instructor hides his/her local R packages. If you need specific R packages for your course, submit a help ticket, and we will install the package so that all attendees can use the same version.
You can also use RStudio server on Open Ondemand to teach the course. Logon ondemand.htc.crc.pitt.edu, Select Interactive Apps -> RStudio Server 2022
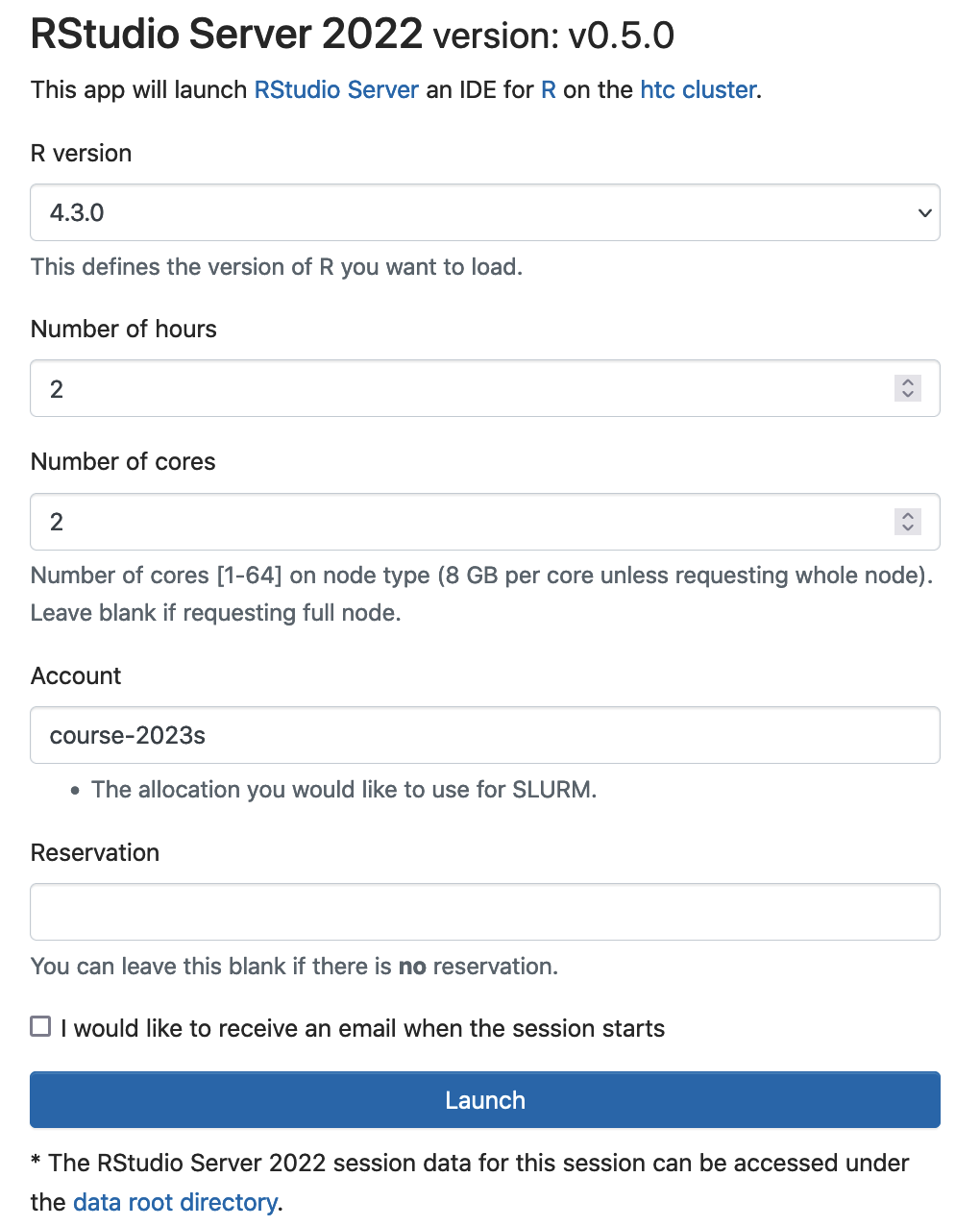
Click Launch to start RStudio server. Slurm will submit a batch job to request 2 cores (16 GB memory), 2 hour walltime. The RStudio server will be run by the login user and the rsession will run the specified R version. The SUs will be extracted from the slurm account course-2023s.
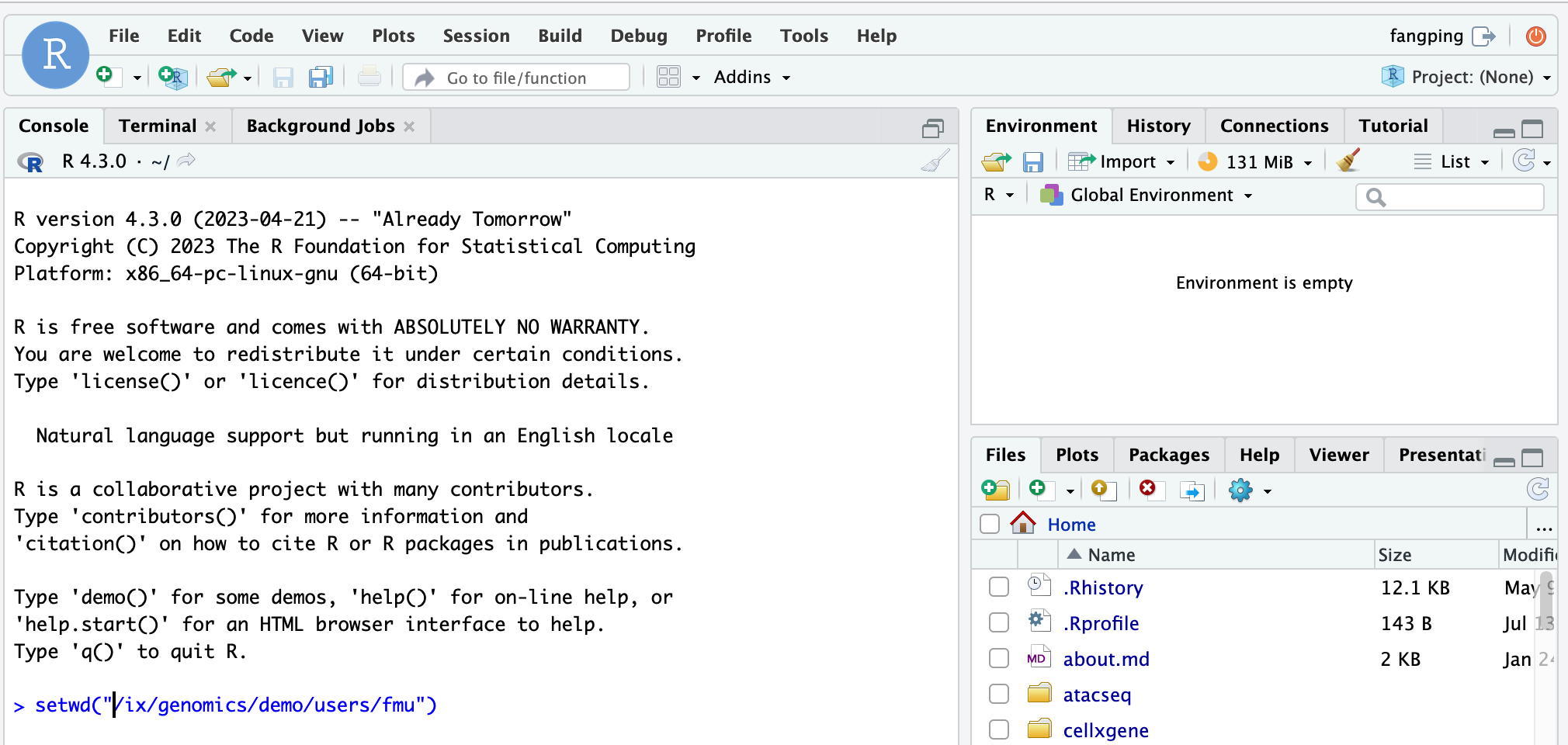
By default, the working directory of the R session is the home directory. You can use setwd() to change the working directory. For example, you can guide each student to change the working directory as shown above.
The instructor should mkdir the parent folder /ix/genomics/demo/users and chmod it to be group writable.
[fmu@login0b ~]$ mkdir -p /ix/genomics/demo/users [fmu@login0b ~]$ chmod 770 /ix/genomics/demo/users
The instructor can guide each student to create his/her own folder under the course storage. Each student can logon ondemand.htc.crc.pitt.edu, click Clusters -> >_HTC Shell Access
[fmu@login0b ~]$ mkdir -p /ix/genomics/demo/users/fmu [fmu@login0b ~]$ chmod 700 /ix/genomics/demo/users/fmu
You can open a R markdown file. I have copied pbmc3k_tutorial.Rmd from NGS 2022s workshops.
[fangping@login0b ~]$ cd /ix/genomics/demo/users/fmu [fangping@login0b fmu]$ cp /bgfs/genomics/workshops/2022s/Overview_of_NGS_data_analysis_using_Pitt_ondemand_and_R/seurat/pbmc3k_tutorial.Rmd .
Then click Open File from RStudio server and open pbmc3k_tutorial.Rmd. You can Knit to html to generate html output from the R markdown.
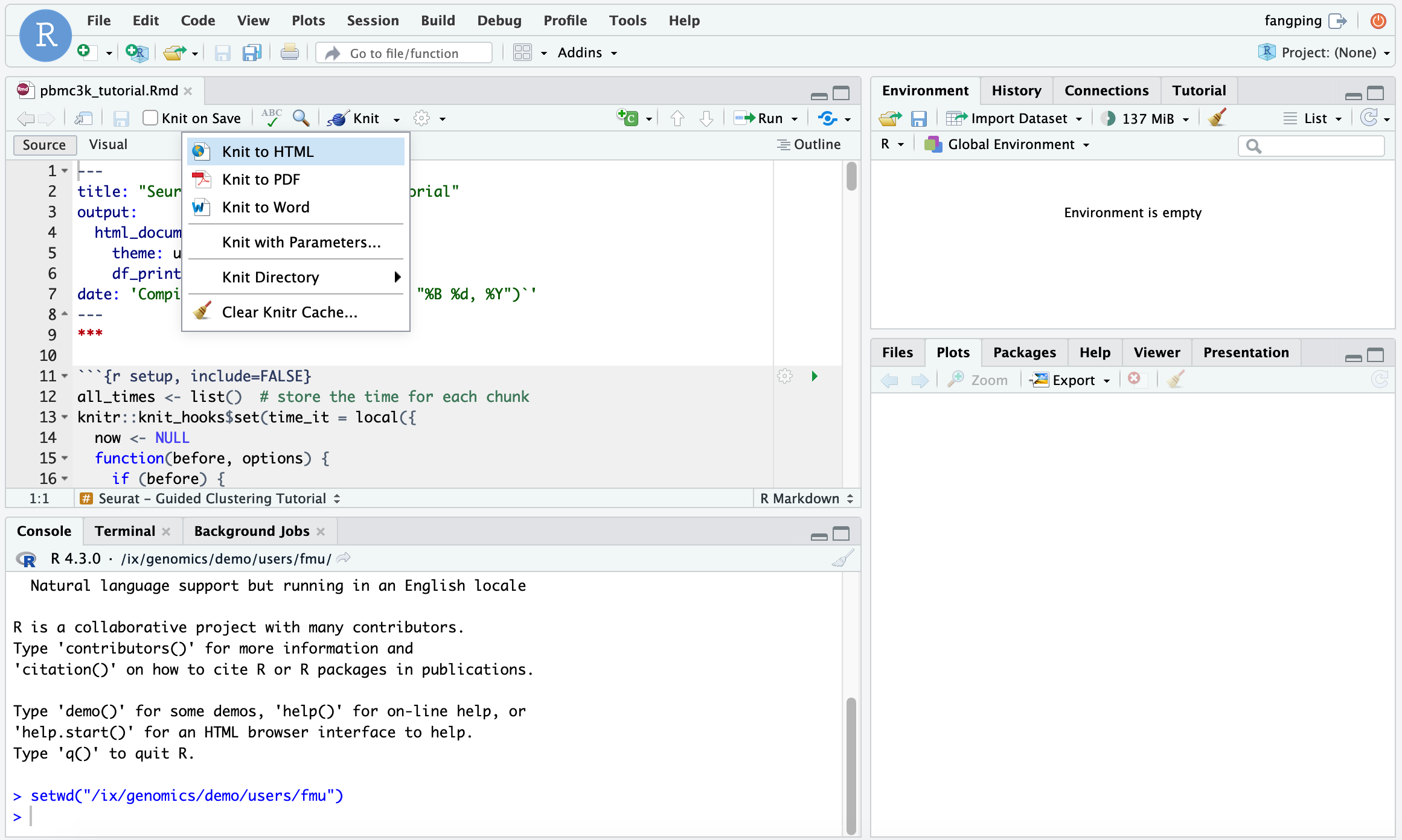
The instructor can also teach students how to submit a R batch job. We recommend that the instructors provide a job template (test.sbatch) as follows.
#!/bin/bash #SBATCH --job-name R_ExampleJob #SBATCH --account=course-2023s # use your course allocation #SBATCH --nodes=1 # request a single node #SBATCH -c 1 # request 1 core #SBATCH --time=01:00:00 # 1 hour walltime # load R module module load gcc/12.2.0 r/4.3.0 #the instructors or students can write the R code in test.R R CMD BATCH test.R test.txt # R CMD BATCH test.R #output will be directed to test.Rout
To submit this job, run "sbatch test.sbatch"
The instructor can also teach other advance topics, including parallel processing or high throughput computing jobs. You can refer to https://crc.pitt.edu/r_and_rstudio
Using conda and python
We have installed multiple anaconda python as modules and these modules can also be used through Open Ondemand Jupyter notebook/lab.
python/ondemand-jupyter-python3.8
python/ondemand-jupyter-python3.9
python/ondemand-jupyter-python3.10
Each anaconda python distribution includes more than 6000 python packages.
You can select the corresponding module, and run jupyter notebook/lab using the anaconda python distribution. Logon ondemand.htc.pitt.edu, Interactive Apps -> Jupyter
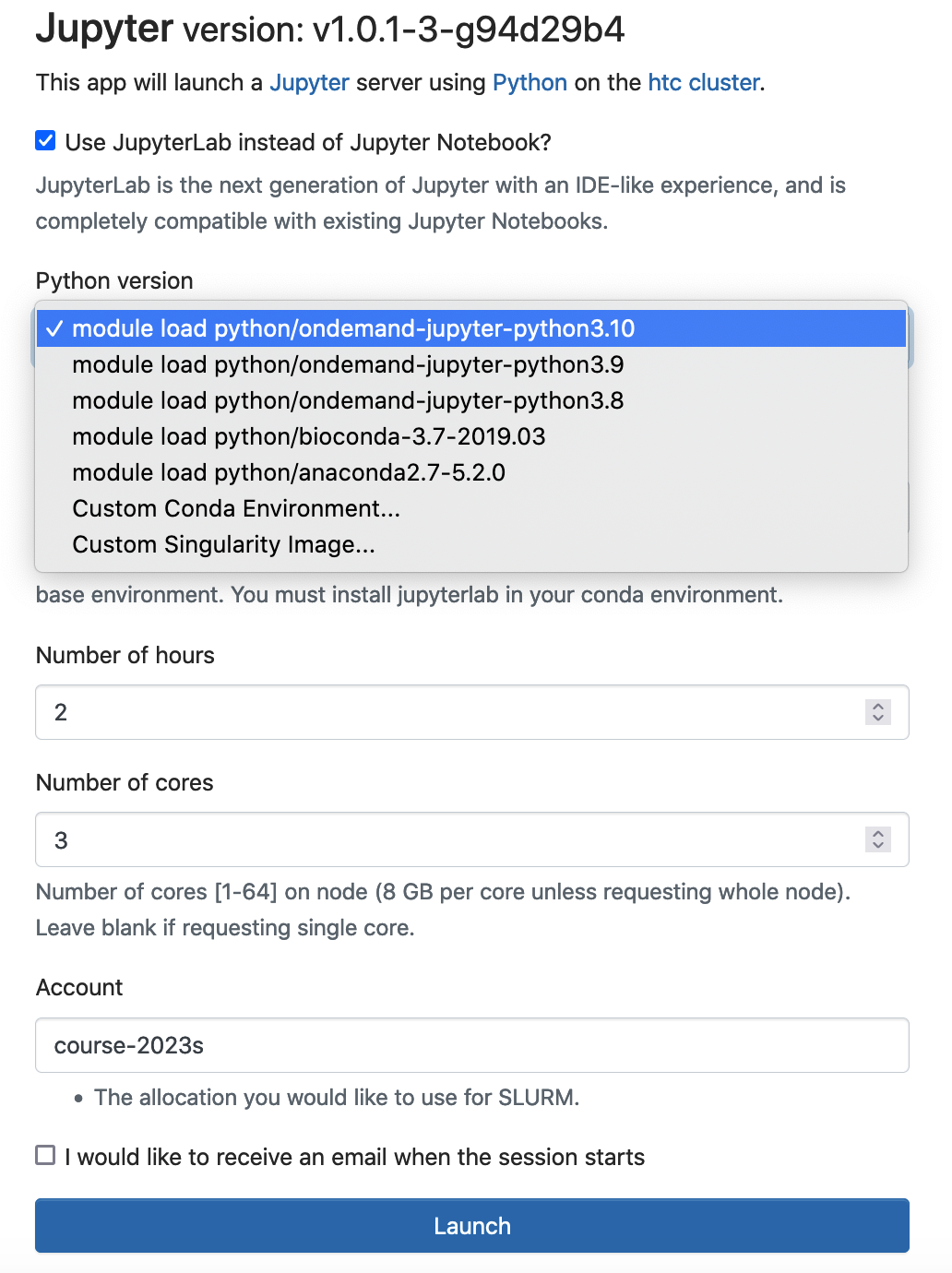
Conda is an open source package management system and environment management system. The instructor can generate conda environment and use conda as a package manager to install, run and update packages and their dependencies. We recommend that the instructor generate conda environment(s) under the course storage. All course attendees should use this conda environment(s).
[fangping@login0b ~]$ cd /ix/genomics/demo/software [fangping@login0b software]$ module load python/ondemand-jupyter-python3.10 [fangping@login0b software]$ conda create --prefix=/ix/genomics/demo/software/env python=3.10 ... [fangping@login0b software]$ source activate /ix/genomics/demo/software/env (/ix/genomics/demo/software/env) [fangping@login0b software]$
DO NOT activate your environment using “conda activate”
Now the instructor can install software packages related to the course to the conda environment.
Bioconda lets you install thousands of software packages related to biomedical research using the conda package manager.
https://bioconda.github.io/recipes/hisat2/README.html
(/ix/genomics/demo/software/env) [fangping@login0b software]$ conda install hisat2
...
To start a Jupyter Lab/notebook with this conda environment activated, you can specify the location of the conda environment under “Name of Custom Conda Environment”.
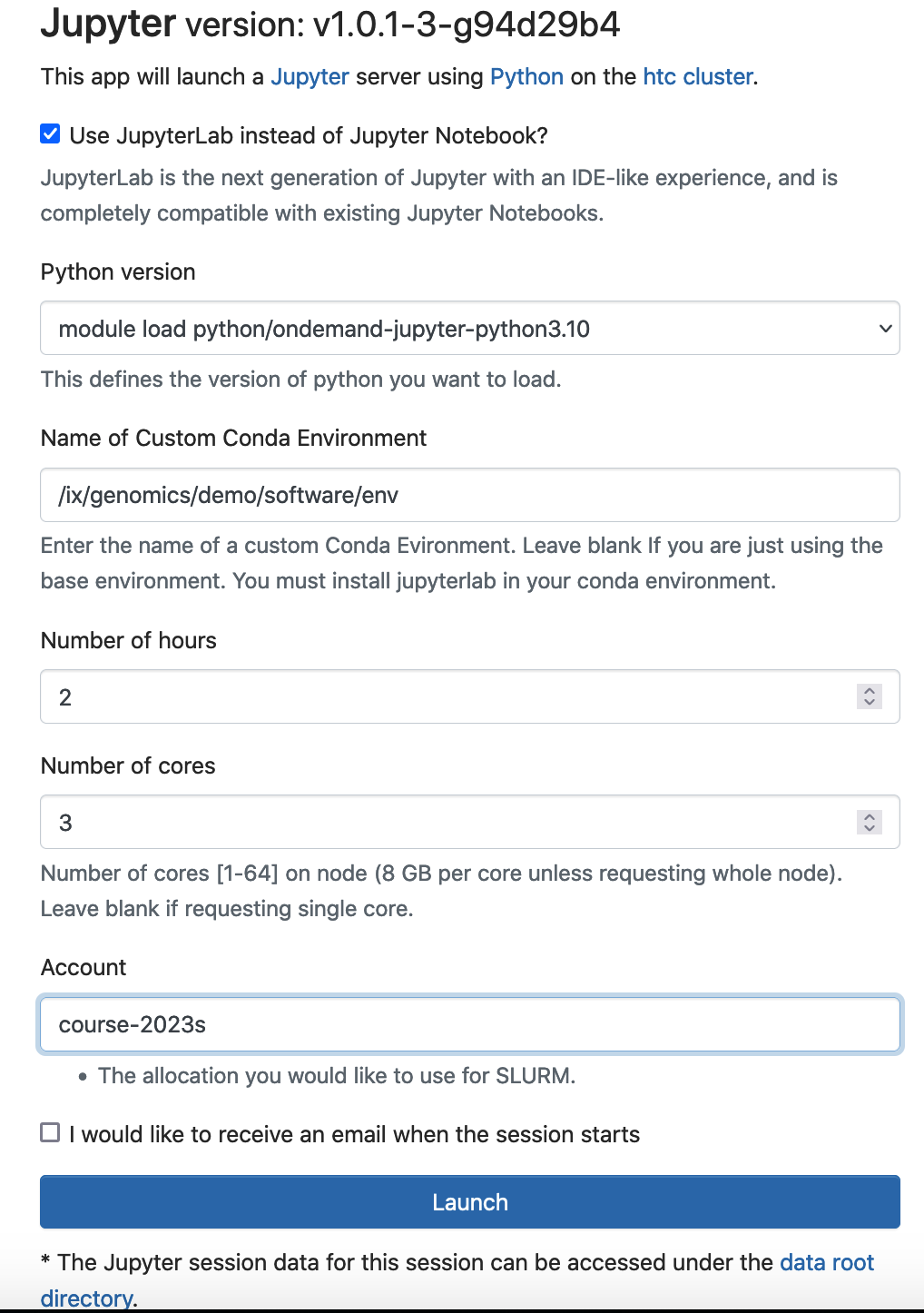
If you receive this error "Failed to connect to htc-n??.crc.pitt.edu:?????" when you Lauch the Jupyter. Wait 1-2 minuates, then refresh your browser.
To use the python packages inside this conda environment, you can directly import the python package. To demonstrate the other packages that you have installed in the conda environment, you can run it through jupyter notebook (See the hisat2 example below).
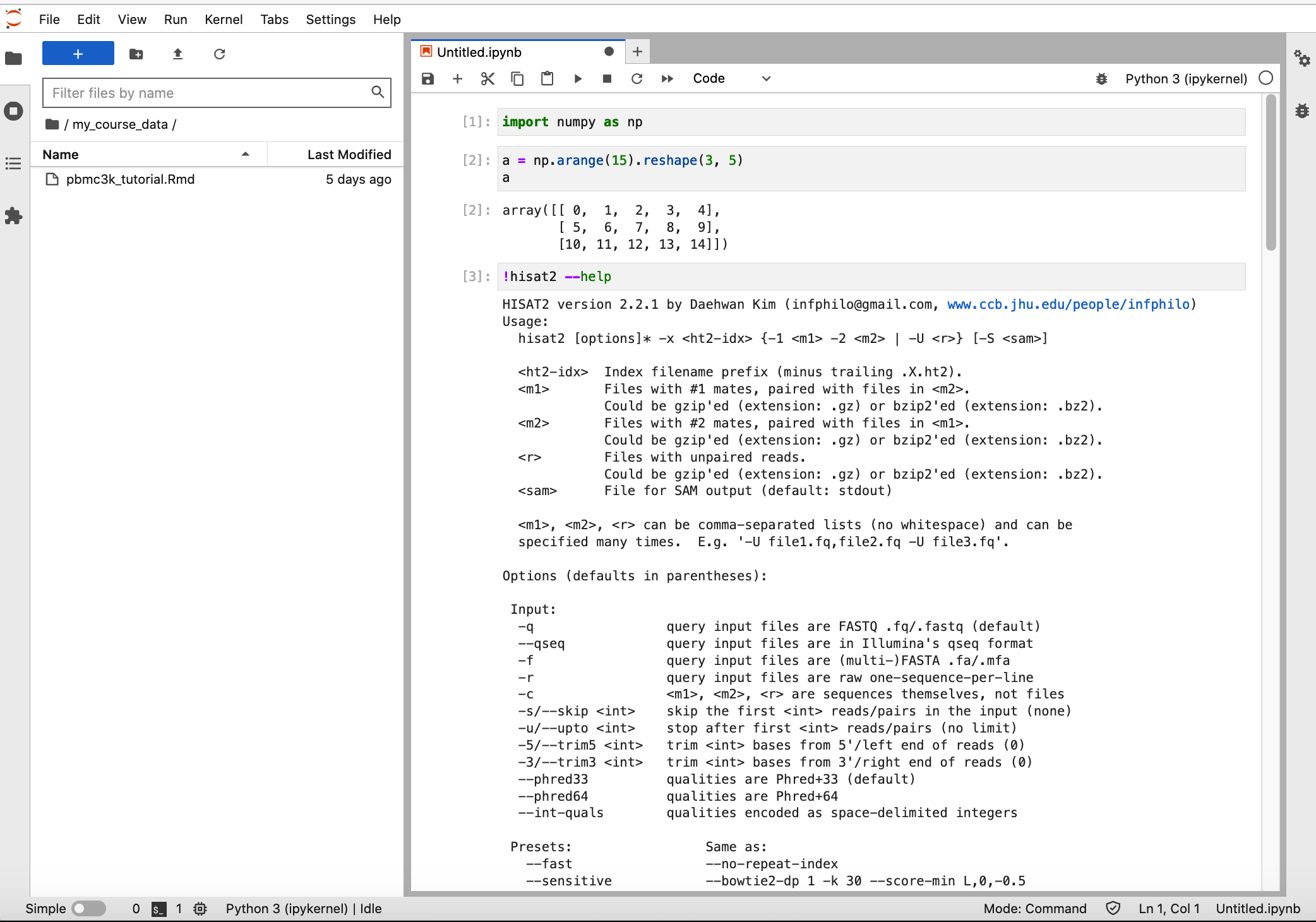
You can also run the other packages through terminal. Launcher -> Terminal
When you start Jupyter Notebook/Lab, the working directory is the home directory. You can use softlink trick to navigate to course storage.
Logon ondemand.htc.crc.pitt.edu, click Clusters -> >_HTC Shell Access
[fmu@login0b ~]$ mkdir -p /ix/genomics/demo/users/fmu # create a folder [fmu@login0b ~]$ chmod 700 /ix/genomics/demo/users/fmu # change the permission [fmu@login0b ~]$ ln -s /ix/genomics/demo/users/fmu my_course_data # generate a softlink to home directory [fmu@login0b ~]$
Each student can navigate to his/her own my_course_data from Jupyter Lab.
The instructor can also teach students how to submit a batch job using the conda environment or other CRC modules. We recommend that the instructors provide a job template (test.sbatch) as follows.
#!/bin/bash #SBATCH --job-name ExampleJob #SBATCH --account=course-2023s # use your course allocation #SBATCH --nodes=1 # request a single node #SBATCH -c 1 # request 1 core #SBATCH --time=01:00:00 # 1 hour walltime # use the custom conda environment module load python/ondemand-jupyter-python3.10 source activate /ix/genomics/demo/software/env # You can also use other modules # module load hisat2/2.2.1 hisat2 --help # run your commands
To submit this job, run "sbatch test.sbatch"