nf-core genomics pipelines
Attention: You are viewing a page from the old CRC user manual.
This page may not represent the most up to date version of the content.
You can view it in our new user manual here.
Nextflow is a reactive workflow framework and a programming DSL that eases the writing of data-intensive computational pipelines.
- Programming language to handle computational workflows, based on Groovy/Java
- Well suited for complex and highly parallel Bioinformatics workflows
- Easy to run/use
- Handles interaction with compute infrastructure, runs just about anywhere
nf-core is a community effort to collect a curated set of analysis pipelines built using Nextflow.
I built a web application through open ondemand:
- Choose a pipeline
- Fill the launch form for that pipeline.
- Launch the pipeline with your choices to the HTC cluster
Point your browser to https://ondemand.htc.crc.pitt.edu
1. Click Genomics Apps -> nf-core pipelines
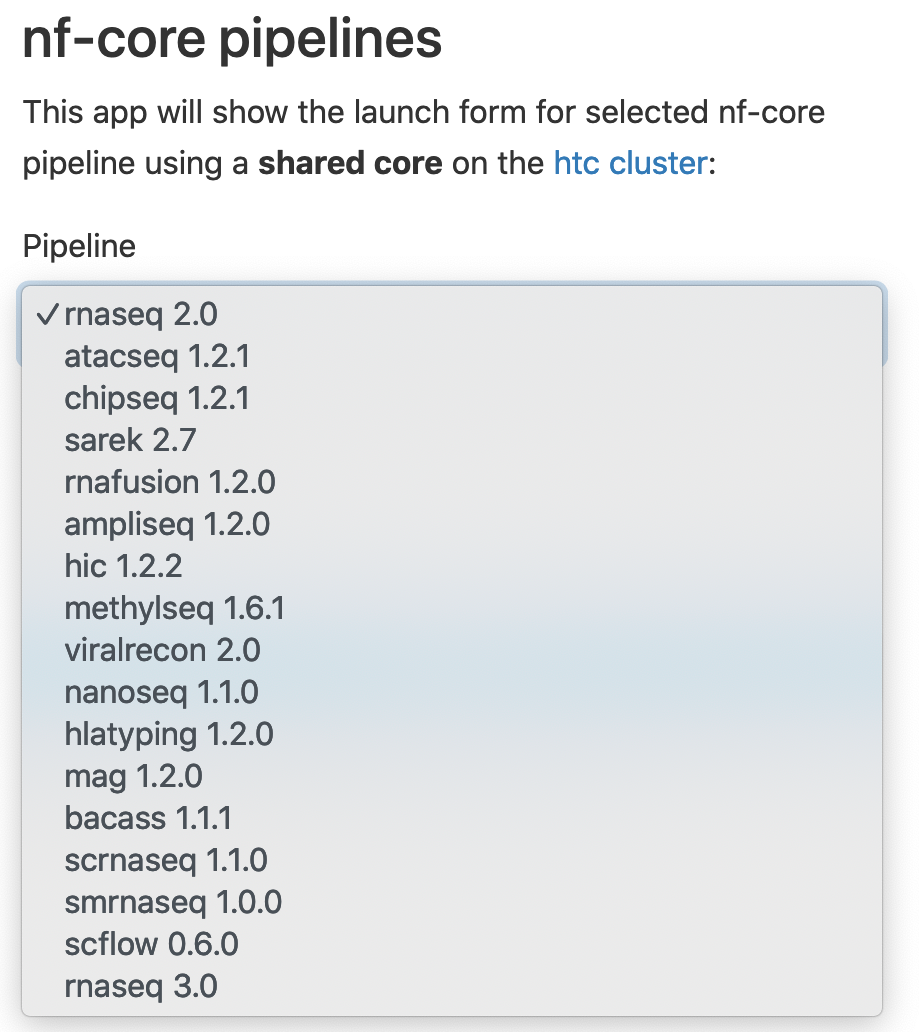
2. Choose a pipelne, for example, rnaseq 2.0, click Launch
3. Click Connect to Nextflow
4. Fill the webform, and click "Launch Workflow".
nf-core/rnaseq
RNA sequencing analysis pipeline using STAR, RSEM, HISAT2 or Salmon with gene/isoform counts and extensive quality control.
1. Logon ondemand.htc.crc.pitt.edu, Click Files -> Home Directory, Click Change directory and go to your bgfs or zfs folder. You can go to /bgfs/genomics/fangping/nf-core-rnaseq to see an example.
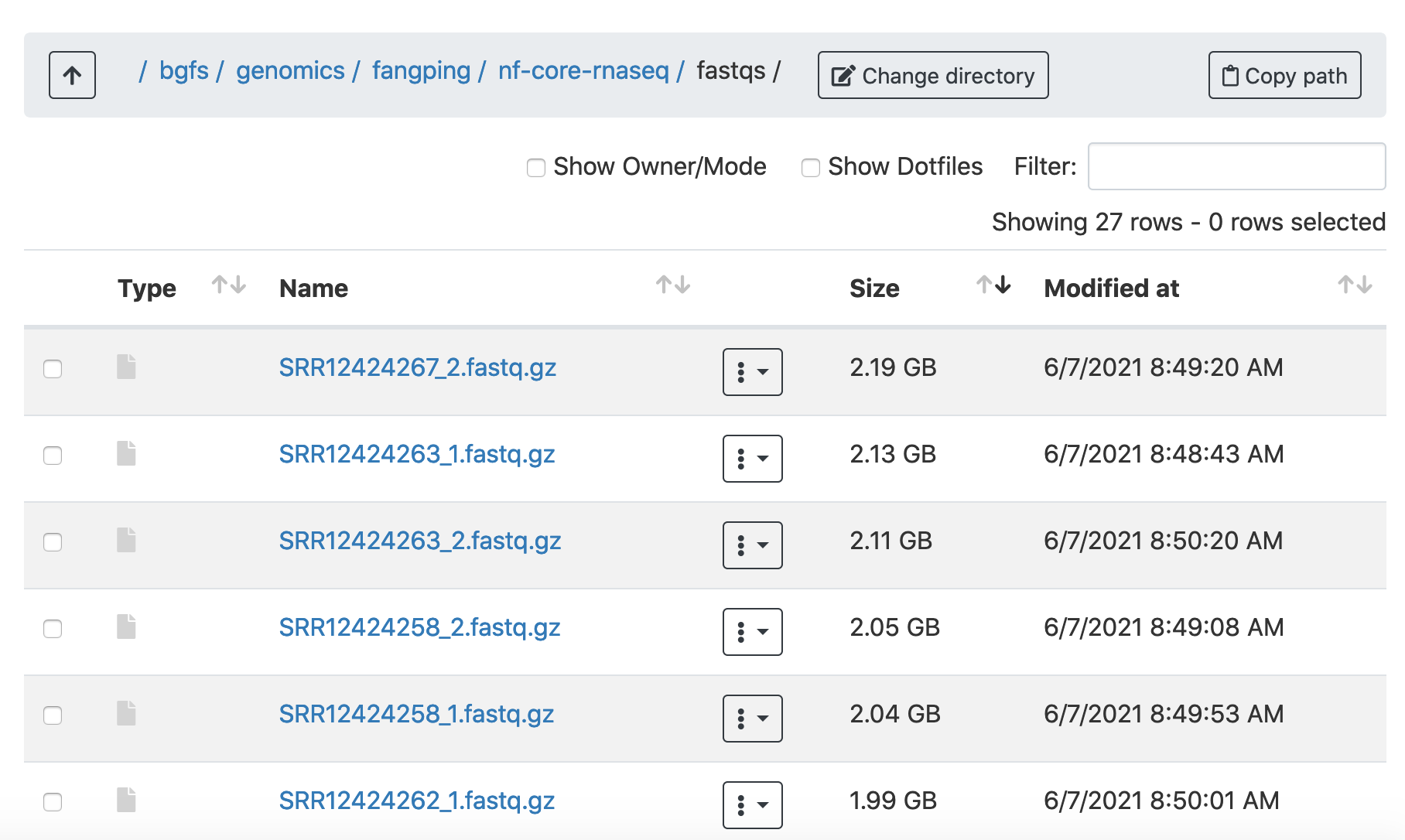
Create a folder fastqs, and transfer your fastqs to this folder. 6 paired-end fastqs from GSE155987 have been downloaded to /bgfs/genomics/fangping/nf-core-rnaseq/fastqs.
Differential Gene Expression Analysis Reveals Global LMTK2 Regulatory Network and Its Role in TGF-β1 Signaling. Cruz DF, Mitash N, Mu F, Farinha CM, Swiatecka-Urban A. Front Oncol. 2021 Mar 18;11:596861. doi: 10.3389/fonc.2021.596861.
The raw fastqs files are within this folder.
2. Samplesheet input
You will need to create a samplesheet file with information about the samples in your experiment before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with 5 columns, and a header row specified in this documentation.
You can use editor on Open Ondemand to edit this file.
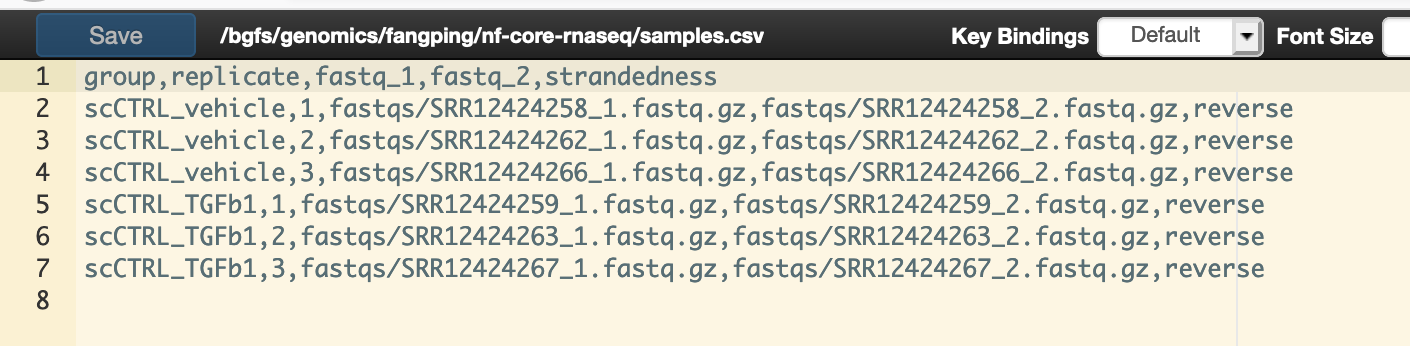
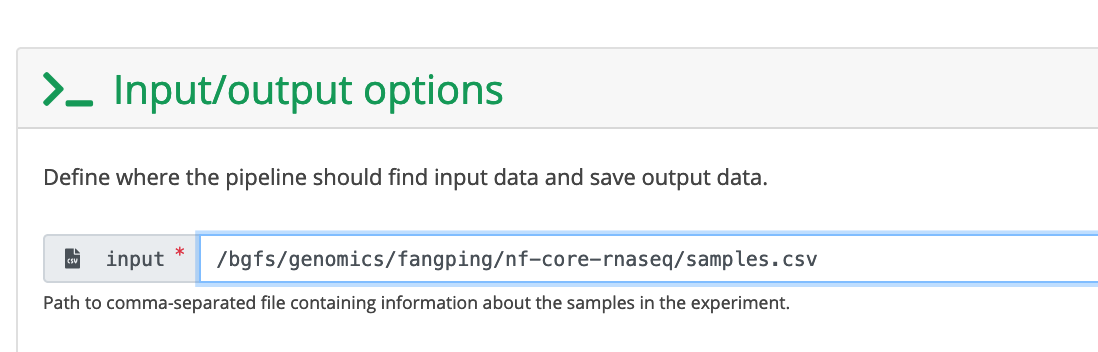
Keep a record of this samplesheet location. You can get its location from the header line of the above editor. For this example, this file is at /bgfs/genomics/fangping/nf-core-rnaseq/samples.csv
3. Reference genome
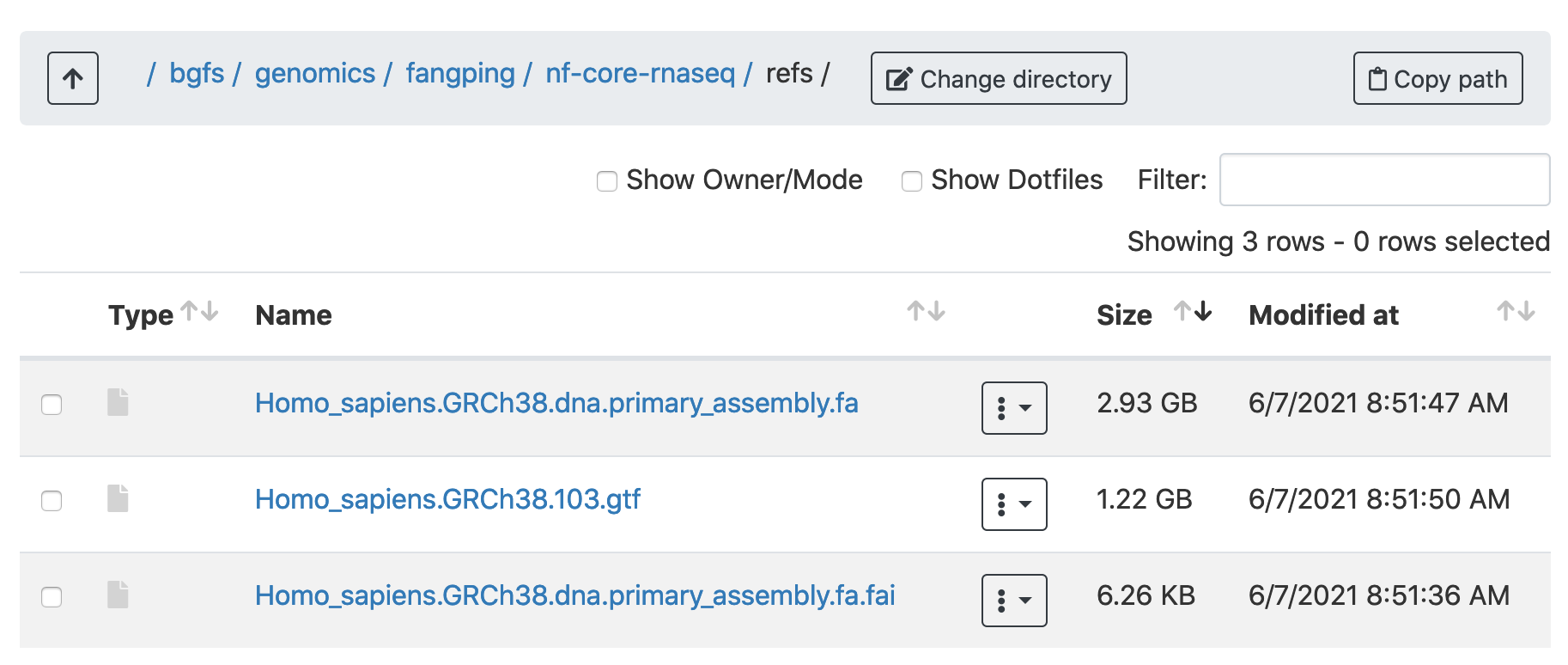
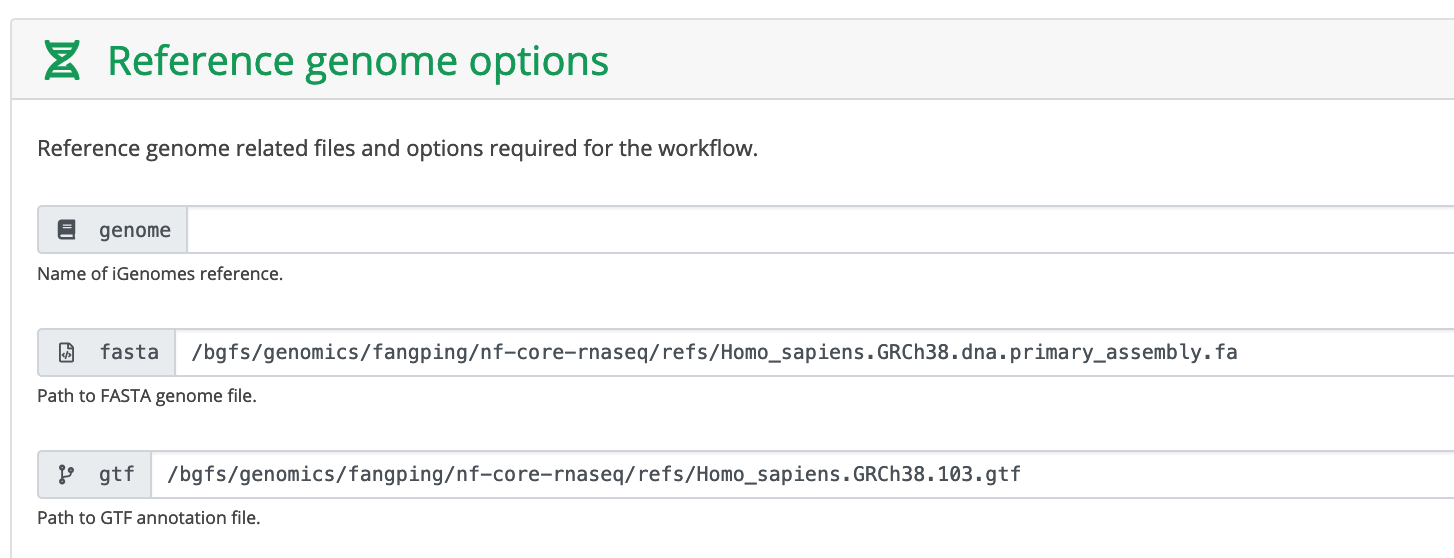
Create a folder refs and download the reference genome for your samples. I have downloaded Ensembl homo sapiens GRCh38 primary assembly and Ensembl annotation version 103 to /bgfs/genomics/fangping/nf-core-rnaseq/refs
4. Fill the launch form
Click Genomics Apps -> nf-core pipelines, choose rnaseq 2.0, click Launch, then click Connect to Nextflow
The input field is the absolute path to your sample sheet.
The minimum reference genome requirements are a FASTA and GTF file, all other files required to run the pipeline can be generated from these files. However, it is more storage and compute friendly if you are able to re-use reference genome files as efficiently as possible. It is recommended to change the save_reference parameter to be true if you are using the pipeline to build new indices.
fasta and gtf fields are the the absolute path to your fasta and gtf file.
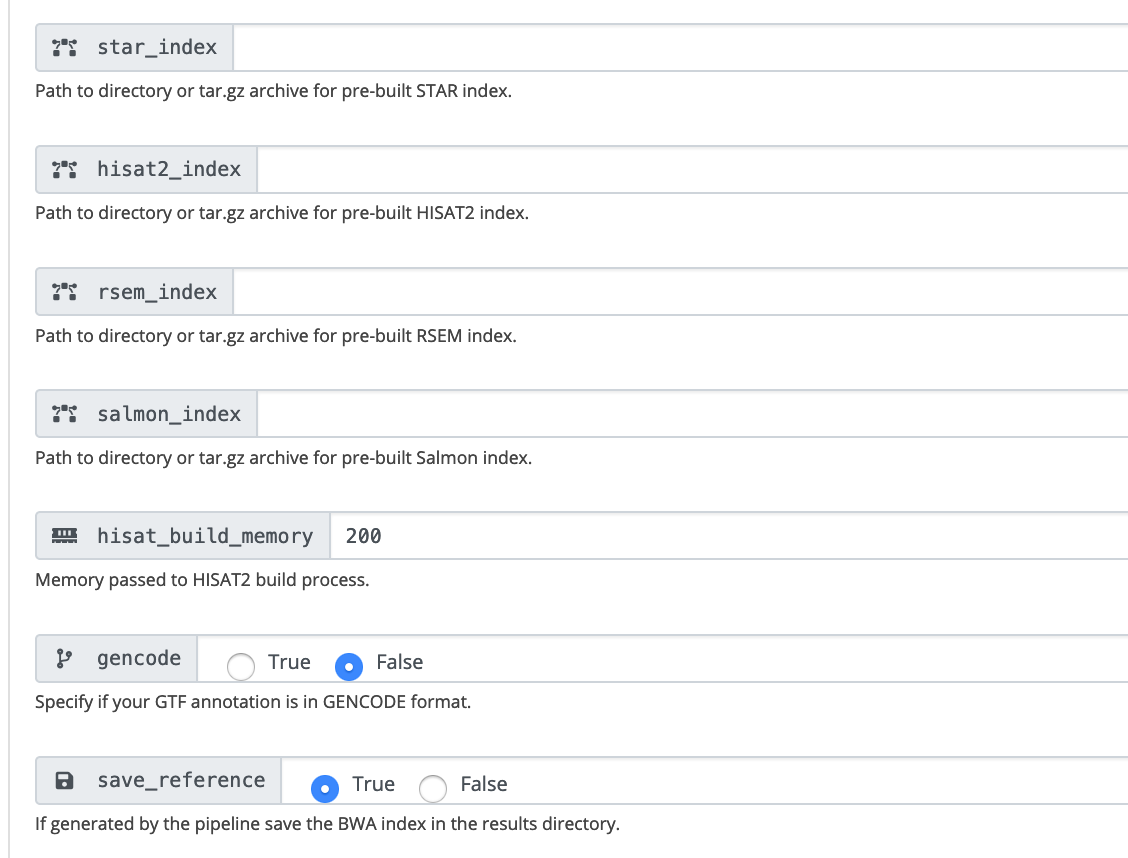
If you are using the pipeline to build new indices, change save_reference to be True. The star index is available at /bgfs/genomics/fangping/nf-core-rnaseq/results/genome/index/star, and you can use this folder as input to star_index for future runs.
Choose the aligner and pseudo_aligner.
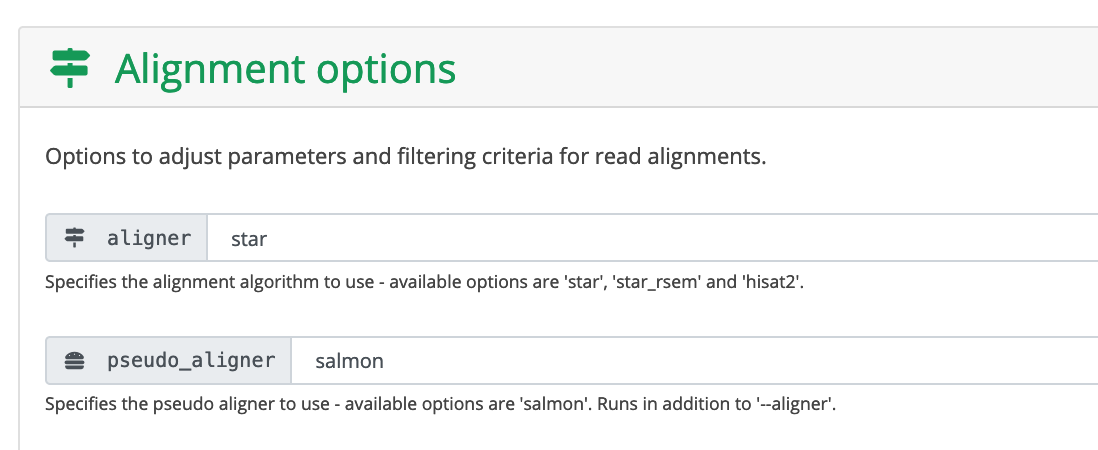
Adjust other parameters based on your data analysis goals. The meanings of these parameters are available here.
5. Lauch workflow
When you are ready, click Launch workflow.
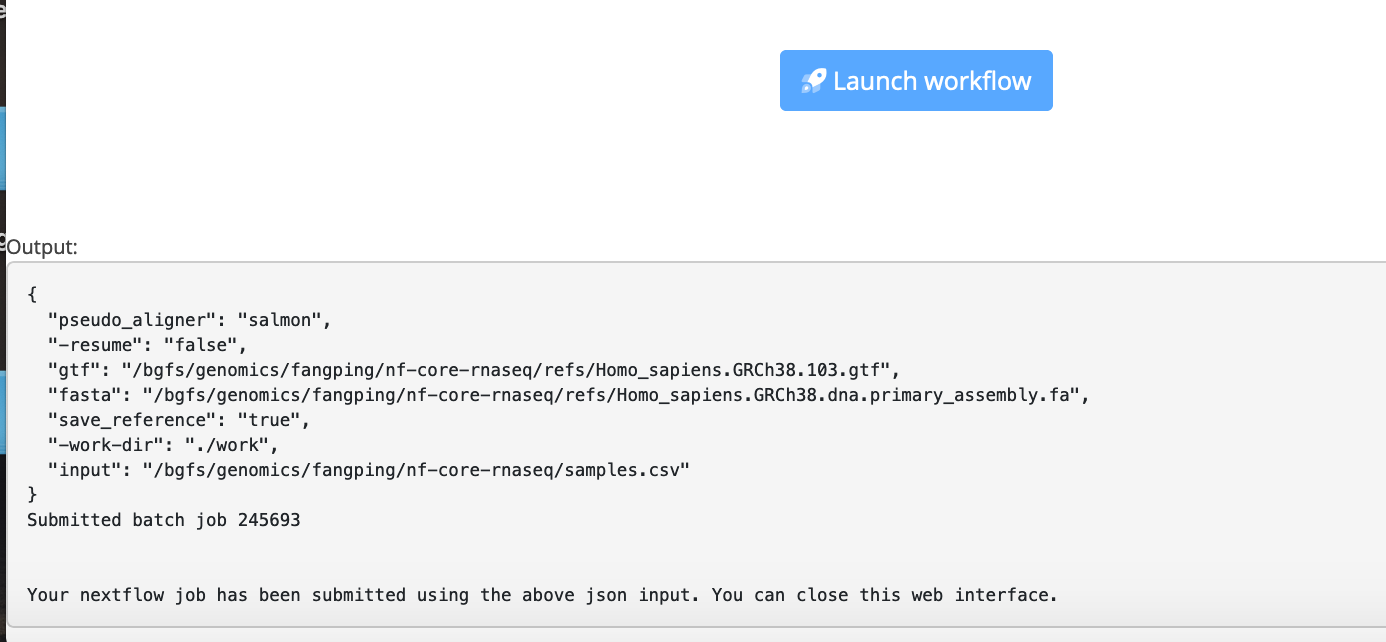
You can close this launch form. You will be notified by emails when your workflow begins and ends.
6. Outputs
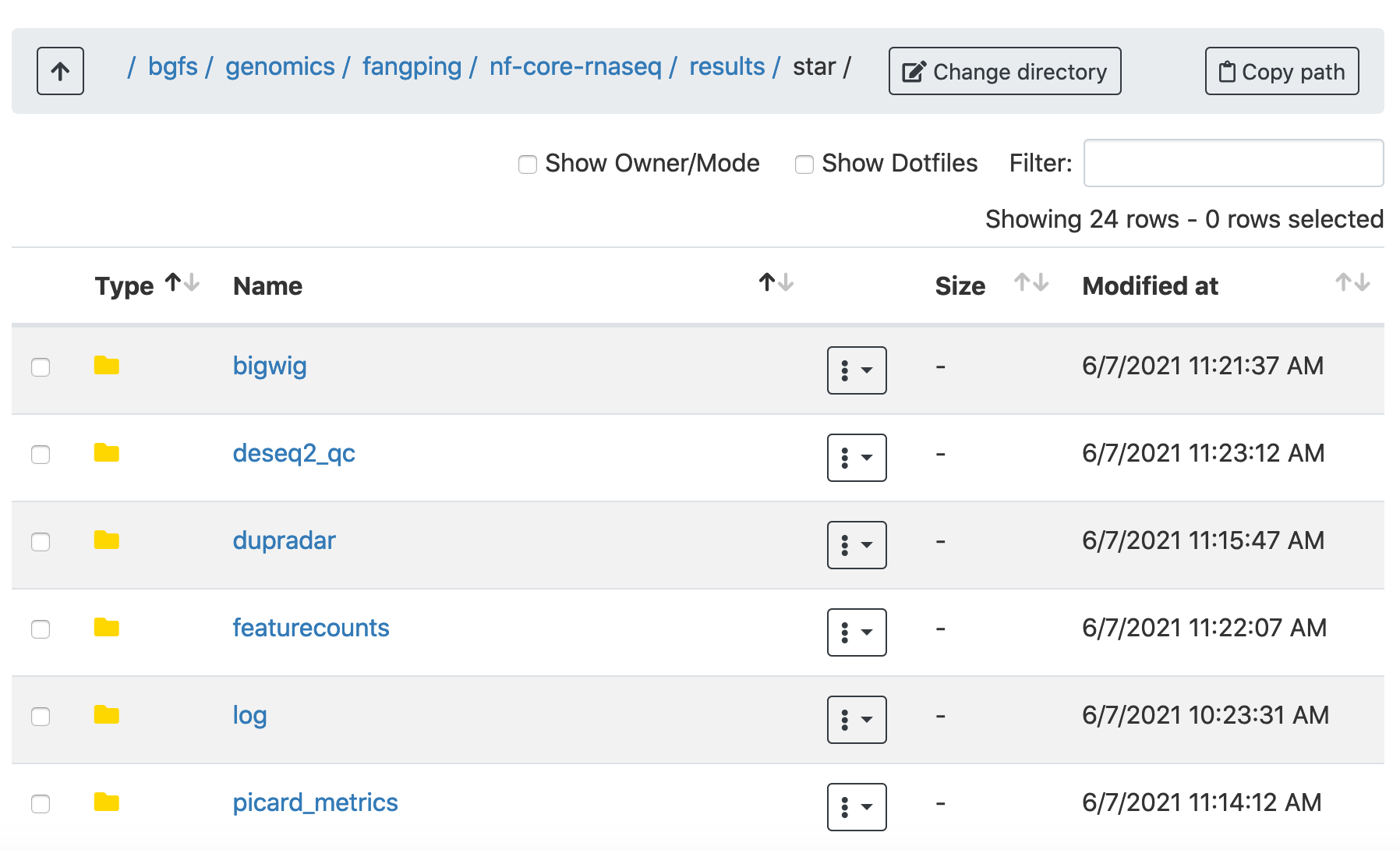
This document describes the output produced by the pipeline. For the above example, featurecounts.merged.counts.tsv is under /bgfs/genomics/fangping/nf-core-rnaseq/results/star/
Navigate to /bgfs/genomics/fangping/nf-core-rnaseq/results/salmon, you can find:
salmon.merged.gene_counts.tsv: Matrix of gene-level raw counts across all samples.
salmon.merged.gene_tpm.tsv: Matrix of gene-level TPM values across all samples.
salmon.merged.transcript_counts.tsv: Matrix of isoform-level raw counts across all samples.
salmon.merged.transcript_tpm.tsv: Matrix of isoform-level TPM values across all samples.
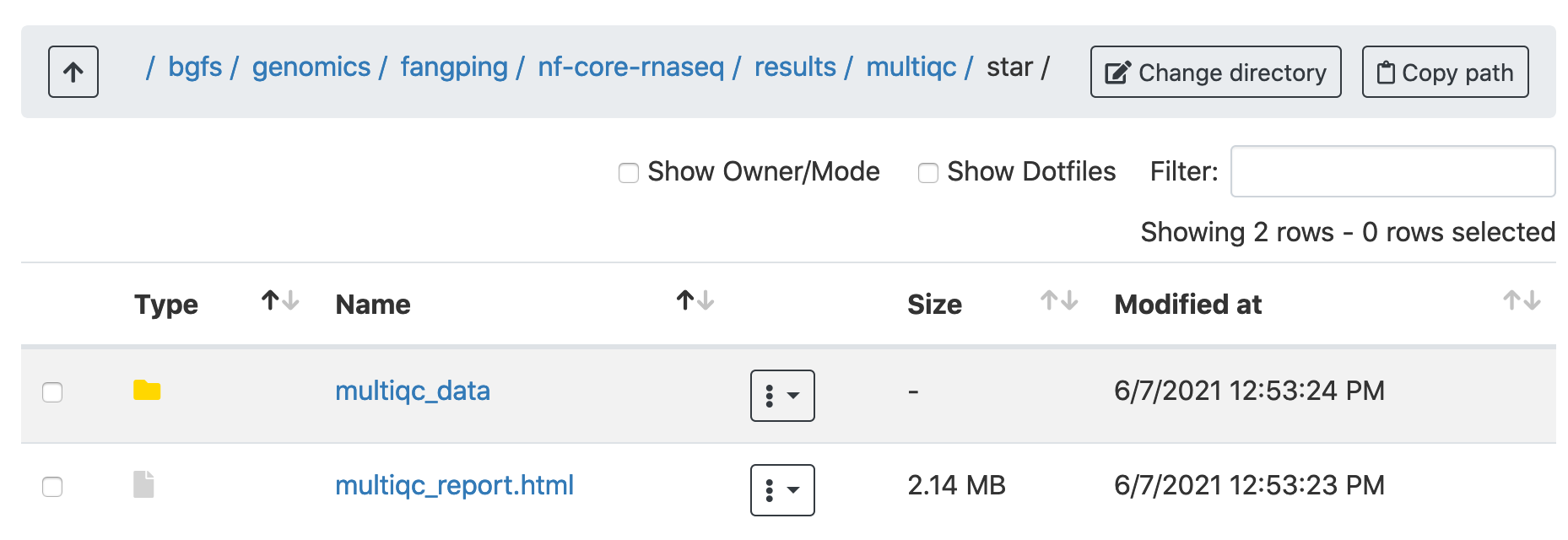
Navigate to /bgfs/genomics/fangping/nf-core-rnaseq/results/multiqc/star, and click multiqc_report.html to open the multiqc results.
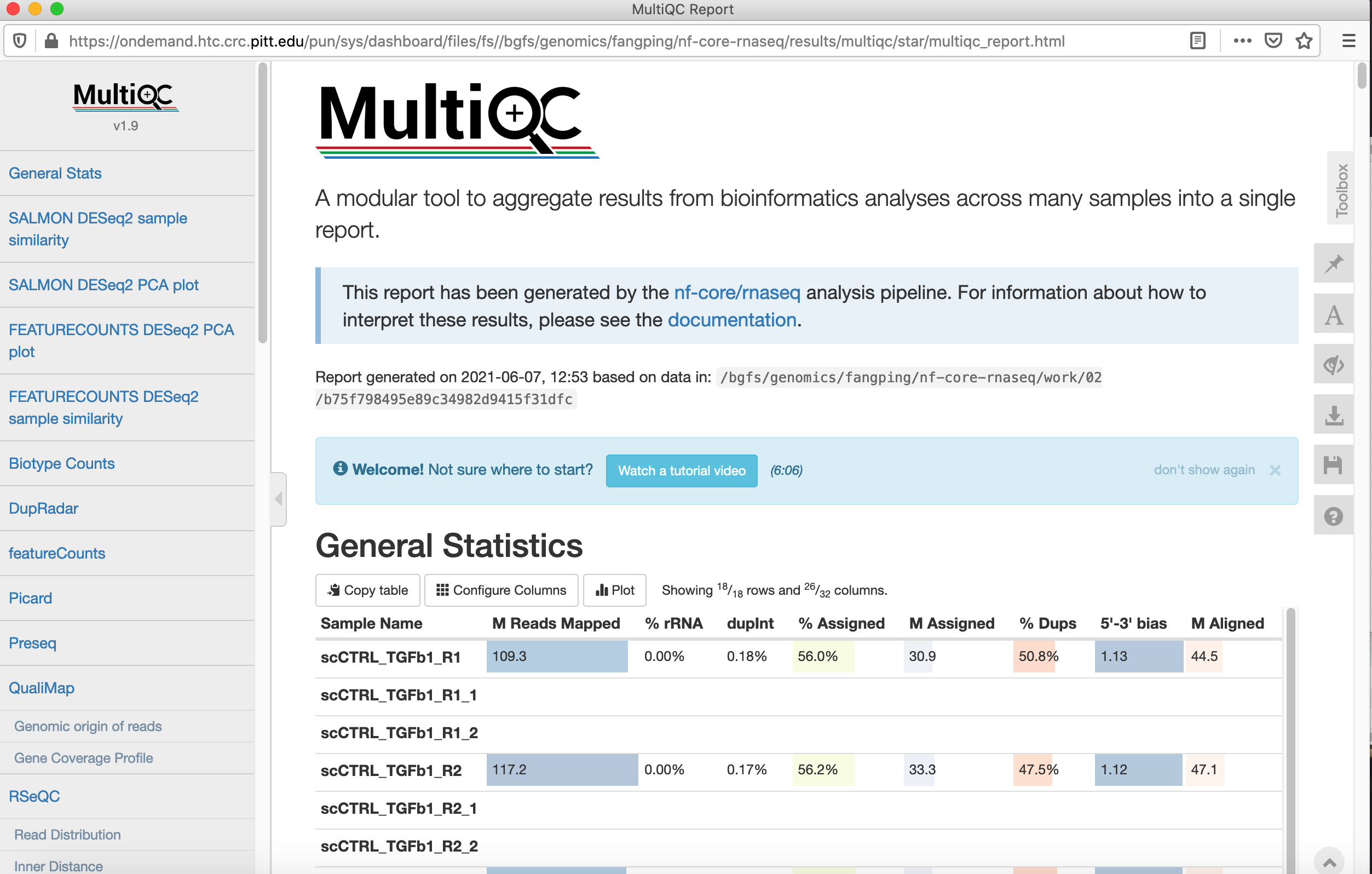
If you prefer to submit jobs through command line iapproach, use /bgfs/genomics/fangping/nf-core-rnaseq/job.sbatch, input.json as reference. Both files are automatically generated through the above web application.
nf-core/sarek
Analysis pipeline to detect germline or somatic variants (pre-processing, variant calling and annotation) from WGS / targeted sequencing. Initially designed for Human, and Mouse, it can work on any species with a reference genome. Sarek can also handle tumour / normal pairs and could include additional relapses.
TODO
nf-core/chipseq
ChIP-seq peak-calling, QC and differential analysis pipeline. nfcore/chipseq is a bioinformatics analysis pipeline used for Chromatin ImmunopreciPitation sequencing (ChIP-seq) data.
1. Logon ondemand.htc.crc.pitt.edu, Click Files -> Home Directory, Click Change directory and go to your bgfs or zfs folder. Test data are in the folder /bgfs/genomics/fangping/nf-core-chipseq/tf and /bgfs/genomics/fangping/nf-core-chipseq/histone
2. H. sapiens single-end ChIP-seq dataset was obtained from 2 separate studies.
Transcription factor data
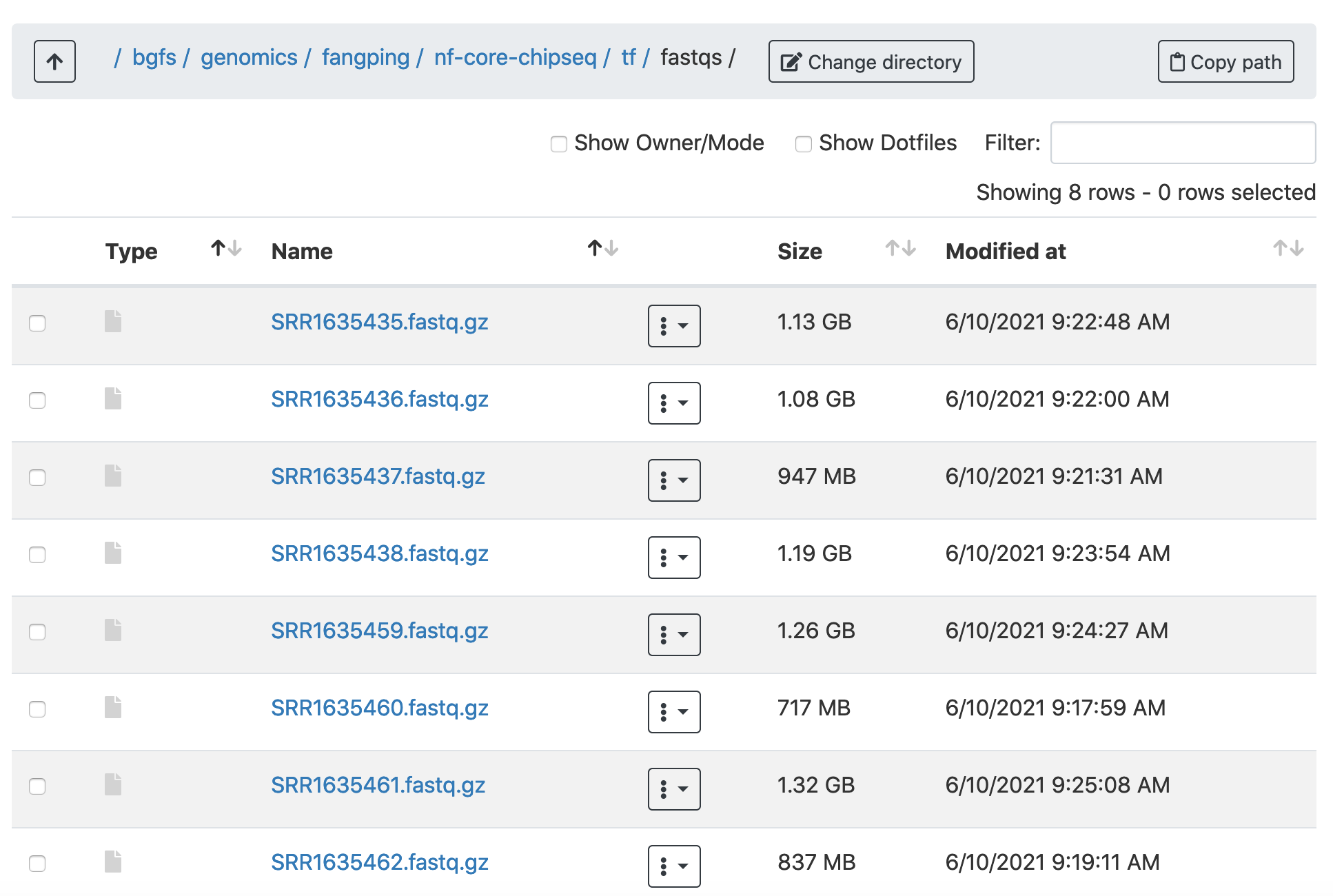
Pubmed, GEO (Input samples: SRR1635435 - SRR1635438, samples: SRR1635459 - SRR1635462)
I have downloaded these data to /bgfs/genomics/fangping/nf-core-chipseq/tf/fastqs
Broad histone data
Pubmed, GEO (samples: SRR1285070 - SRR1285073, Input samples: SRR1285074 - SRR1285077)
I have downloaded these data to /bgfs/genomics/fangping/nf-core-chipseq/histone/fastqs
3.Samplesheet input file
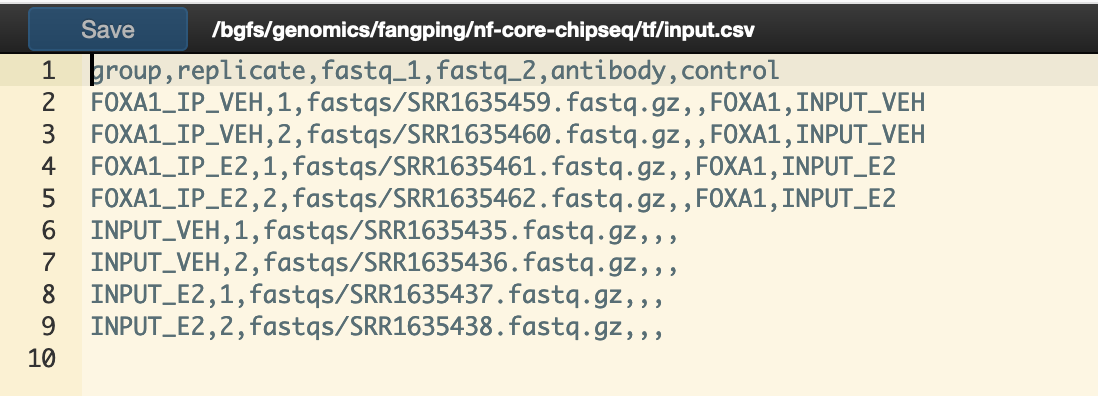
You will need to create a design file with information about the samples in your experiment before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with 6 columns, and a header row contains the following columns.
group,replicate,fastq_1,fastq_2,antibody,control
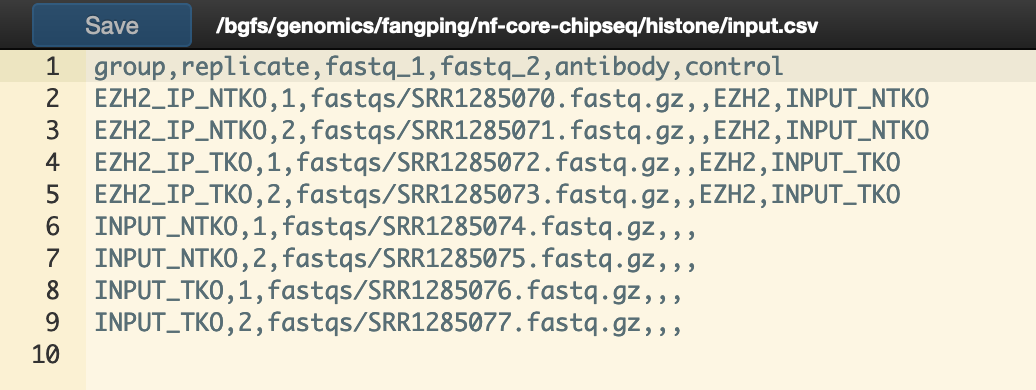
4. Fill the launch form
Click Genomics Apps -> nf-core pipelines, choose chipseq 1.2.1, click Launch, then click Connect to Nextflow
Transcription factor data
The input field is the absolute path to your sample sheet. Choose single_end to be True.
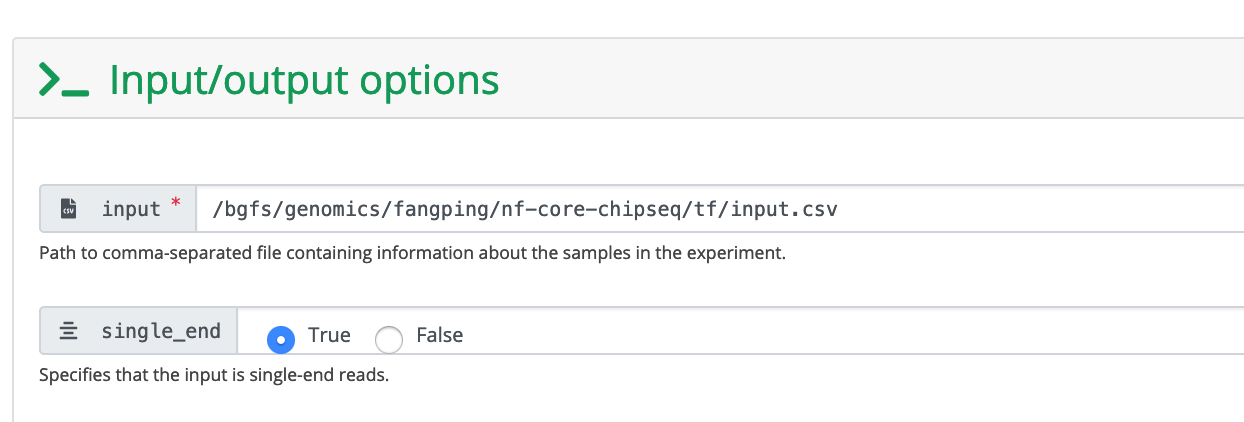
These are human data. We used hg38 as reference genome. You can also provide the absolute path to fasta and gtf files.
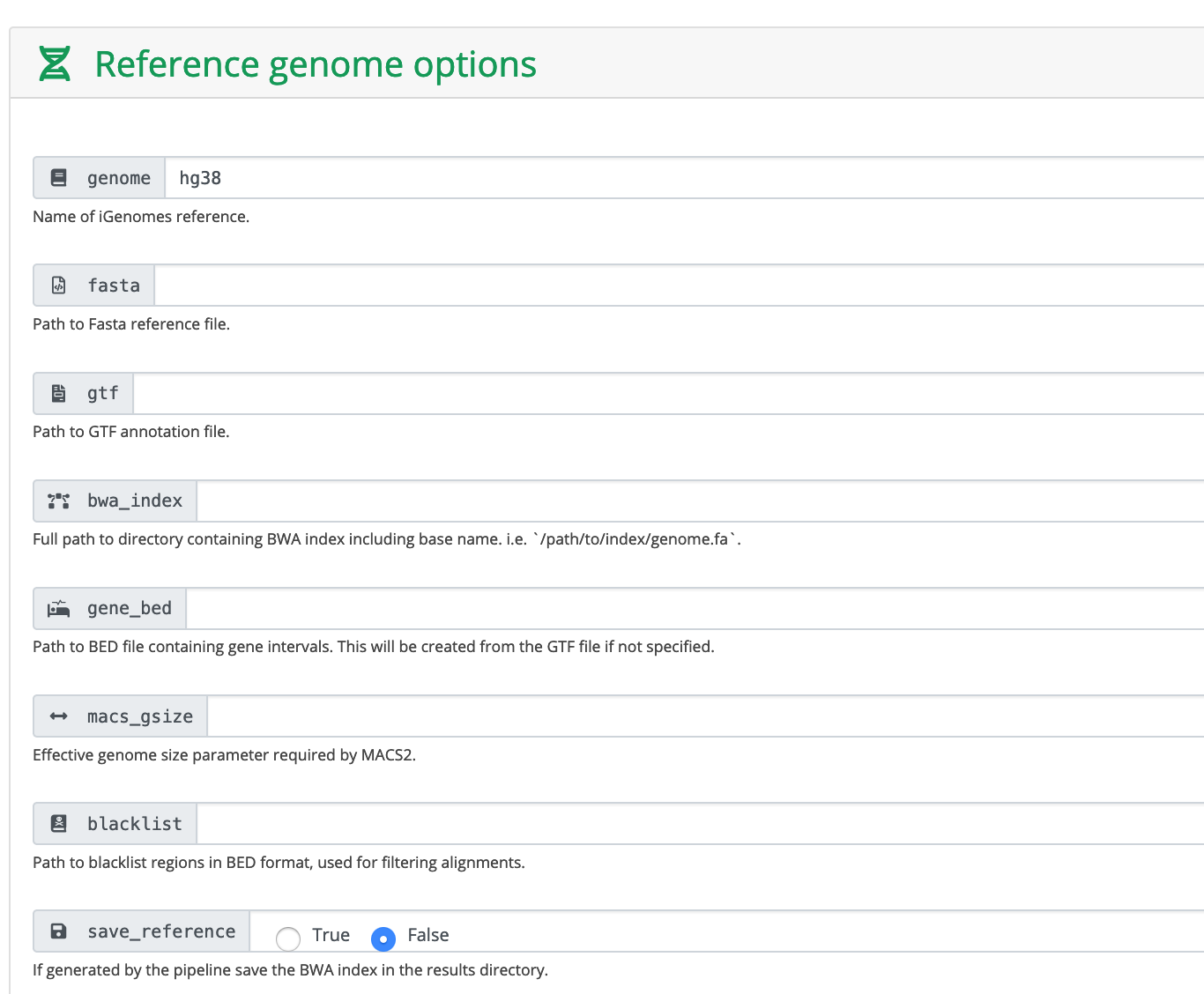
Run MACS2 in narrowPeak mode because these are transcription factor data.
Click Launch workflow.
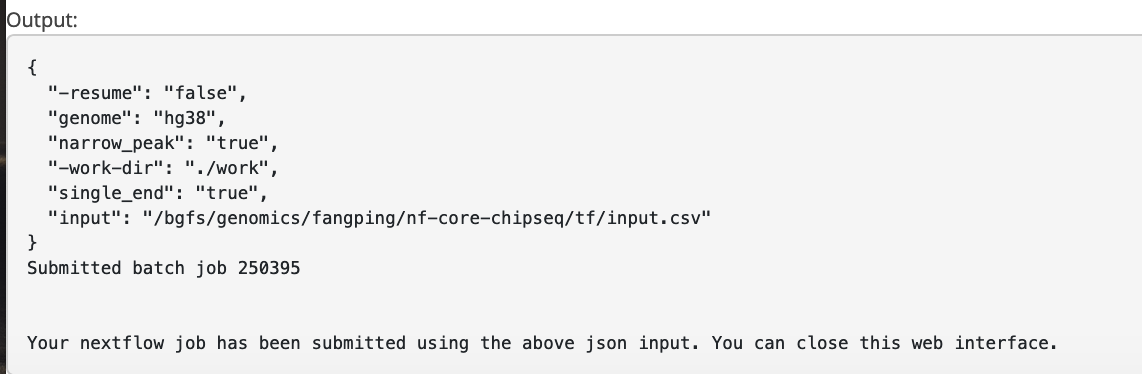
Histone Data
The input field is the absolute path to your sample sheet. Choose single_end to be True.
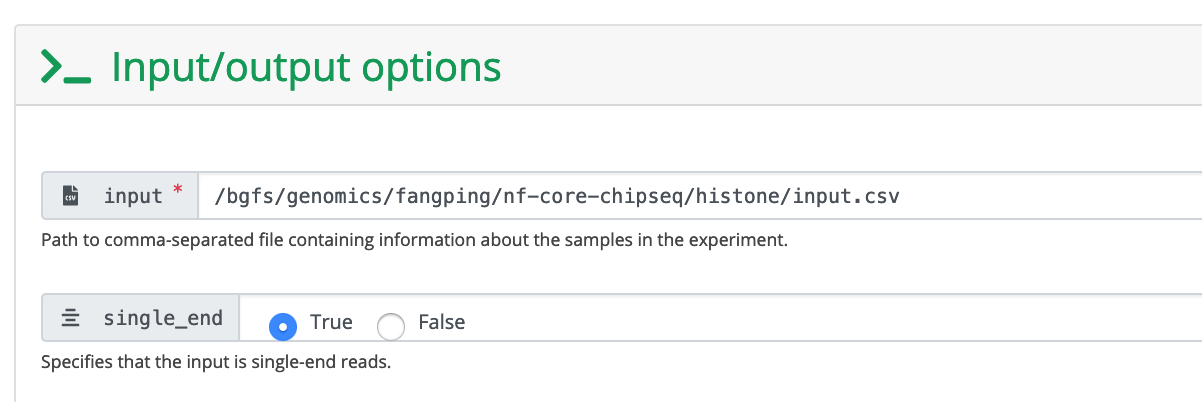
These are human data. We used hg38 as reference genome.
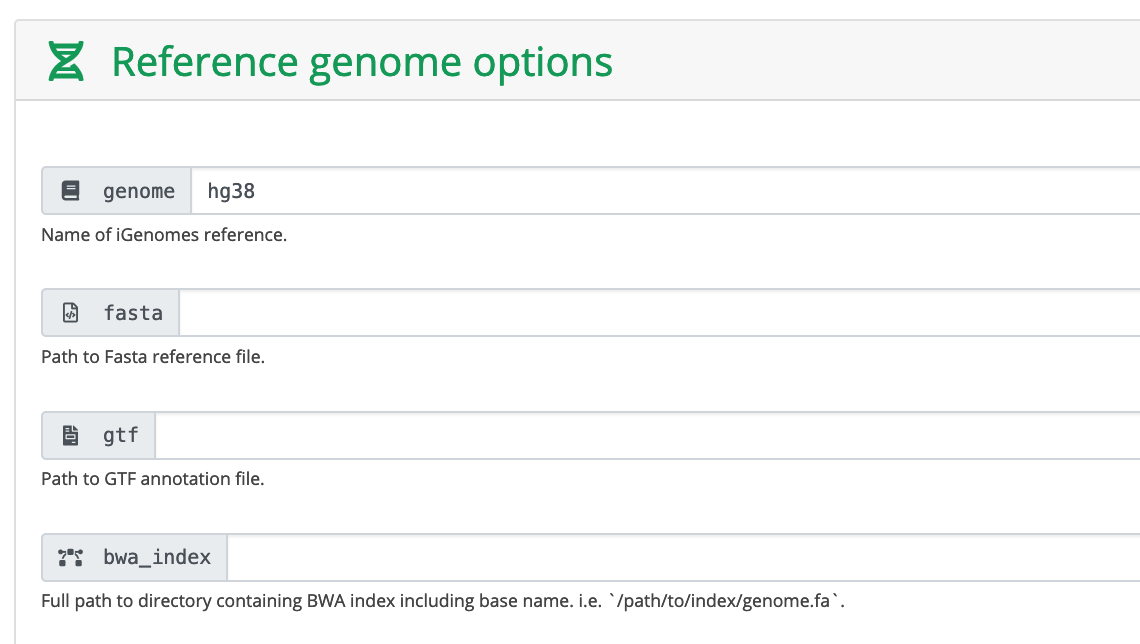
Run MACS2 in broadPeak mode because these are histone data.
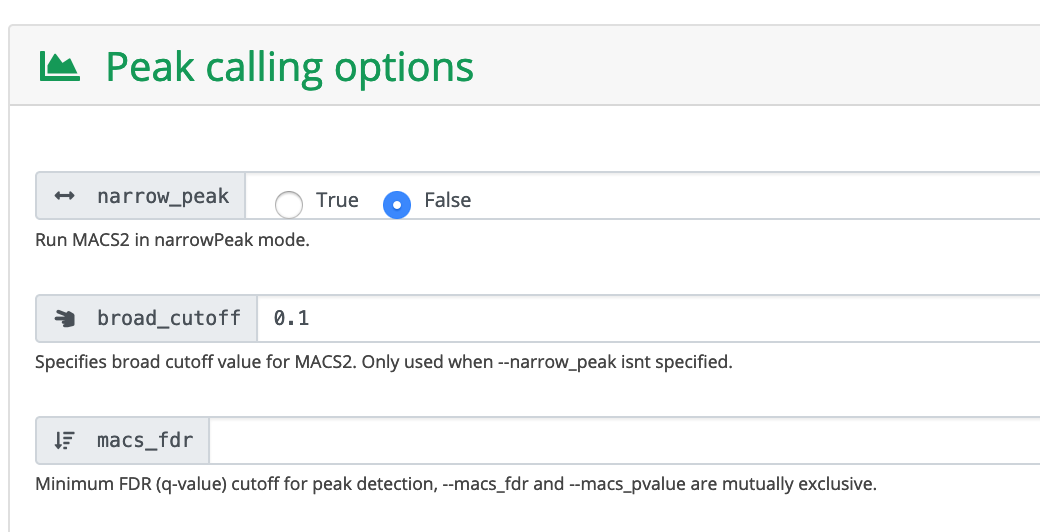
Click Launch workflow.
6. Outputs
This document describes the output produced by the pipeline. Navigate to /bgfs/genomics/fangping/nf-core-chipseq/tf/results and /bgfs/genomics/fangping/nf-core-chipseq/histone/results to read the results.
Navigate to /bgfs/genomics/fangping/nf-core-chipseq/tf/results/multiqc/narrowPeak and /bgfs/genomics/fangping/nf-core-chipseq/histone/results/multiqc/broadPeak to go through the multiqc report.
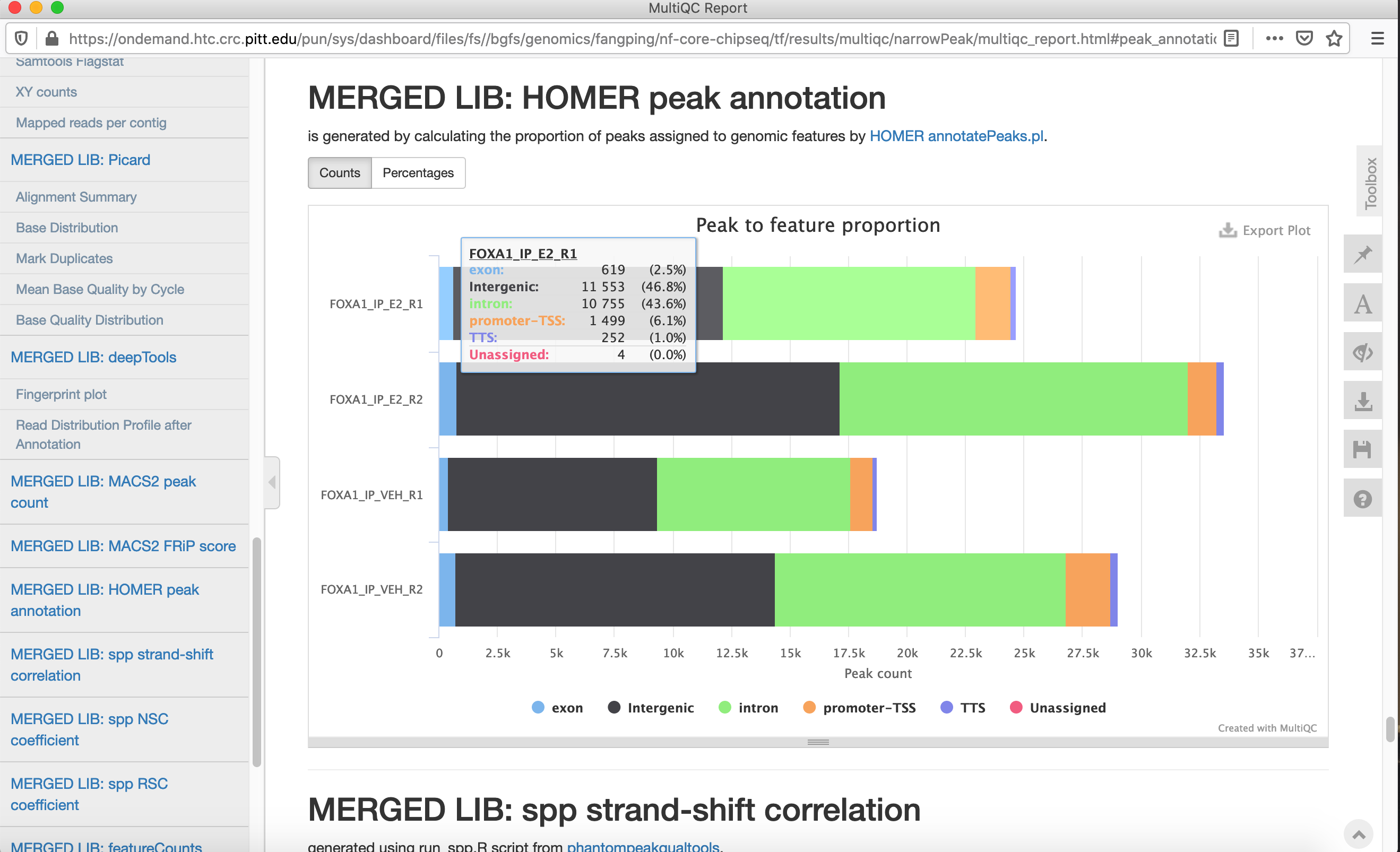
Click multiqc_report.html to open the report.
nf-core/atacseq
ATAC-seq peak-calling, QC and differential analysis pipeline. nfcore/atacseq is a bioinformatics analysis pipeline used for ATAC-seq data.
1. Logon ondemand.htc.crc.pitt.edu, Click Files -> Home Directory, Click Change directory and go to your bgfs or zfs folder. ATACseq test data are in the folder /bgfs/genomics/fangping/nf-core-atacseq
2. Within the fastqs folder, I have downloaded 6 samples from this paper.
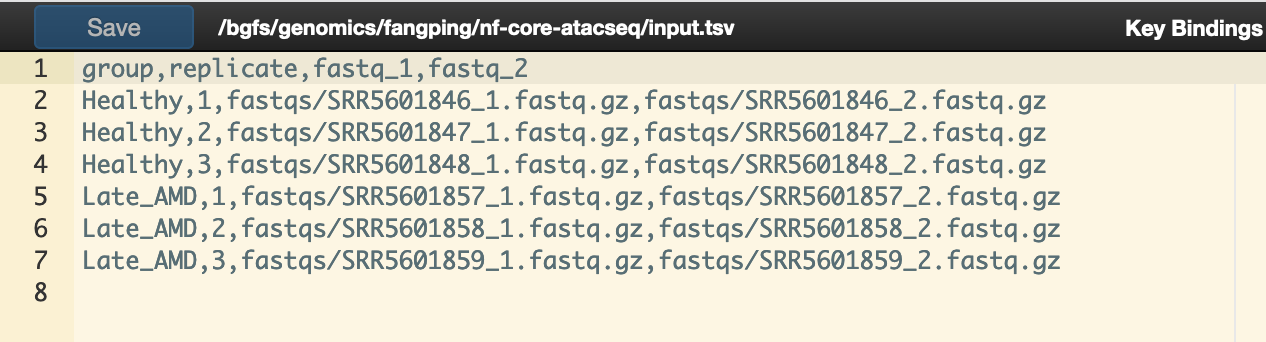
3.Samplesheet input file
You will need to create a design file with information about the samples in your experiment before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with 4 columns.
group,replicate,fastq_1,fastq_2
Example samplesheet is at /bgfs/genomics/fangping/nf-core-atacseq/input.tsv
4. Fill the launch form
Click Genomics Apps -> nf-core pipelines, choose atacseq 1.2.1, click Launch, then click Connect to Nextflow
The input field is the absolute path to your sample sheet.
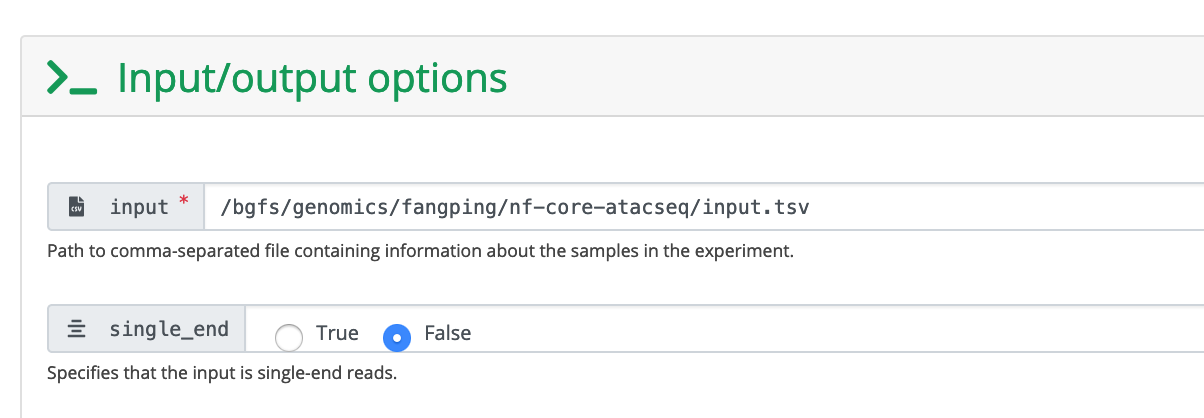
These are human data. We used hg38 as reference genome. You can also provide the absolute path to fasta and gtf files.
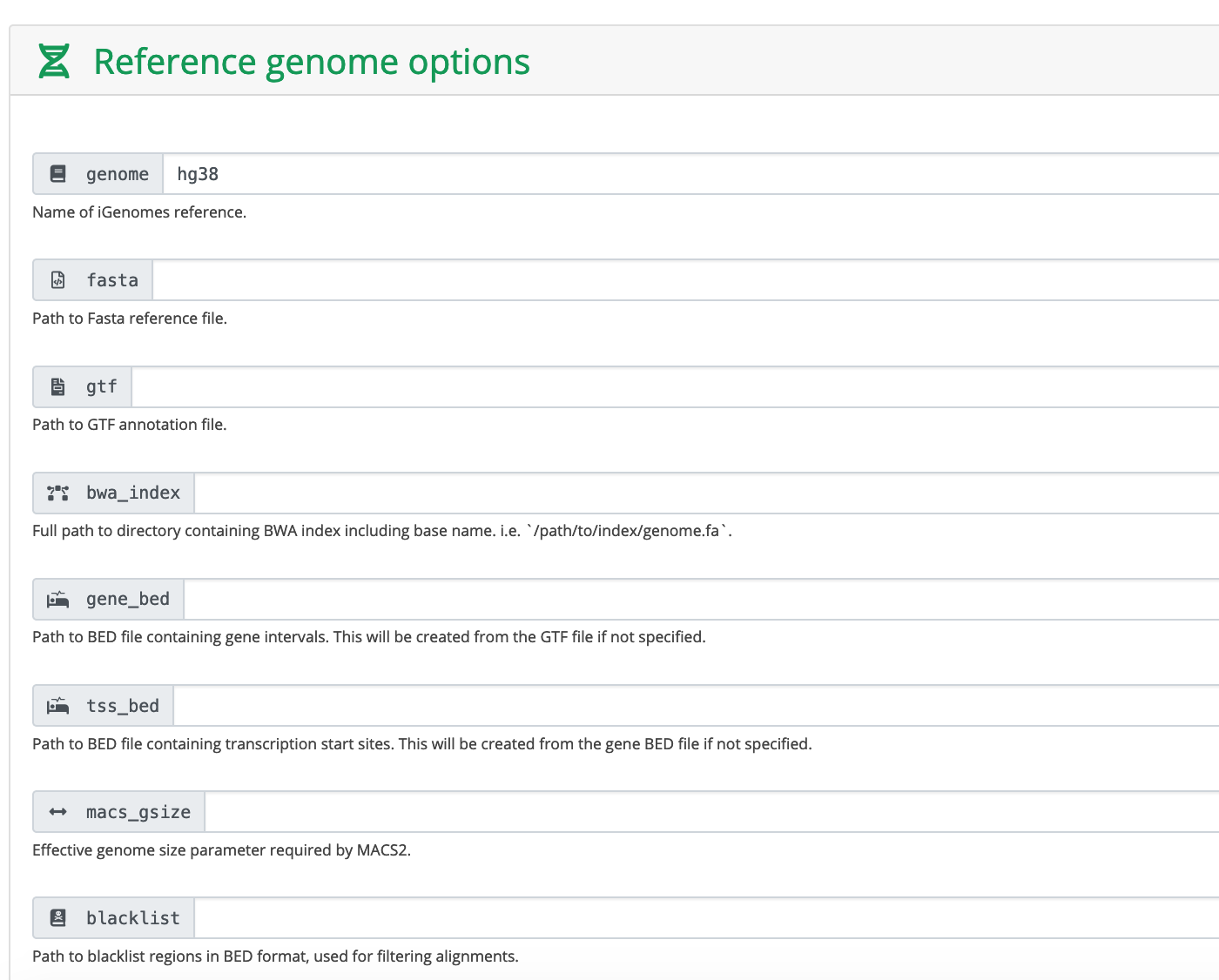
Run MACS2 in narrowPeak mode.
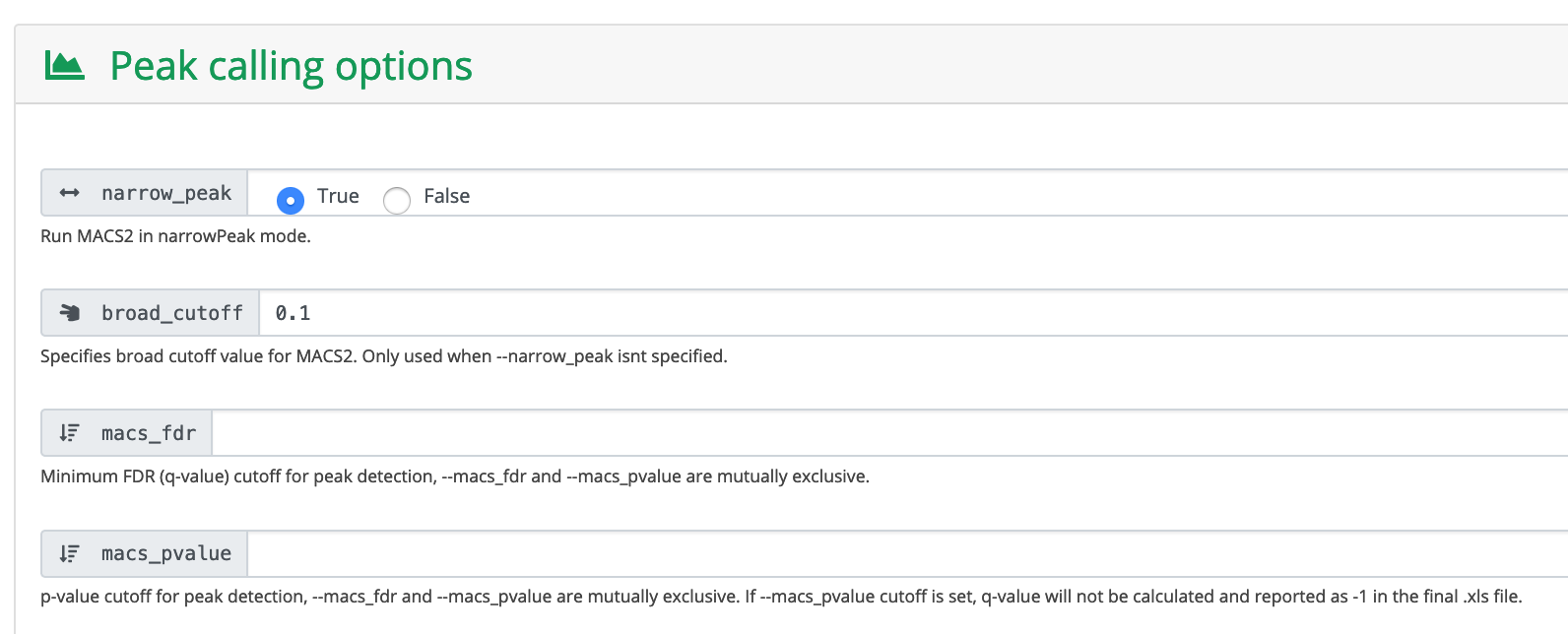
Click Launch workflow.
6. Outputs
This document describes the output produced by the pipeline. Navigate to /bgfs/genomics/fangping/nf-core-atacseq/results to read the results.
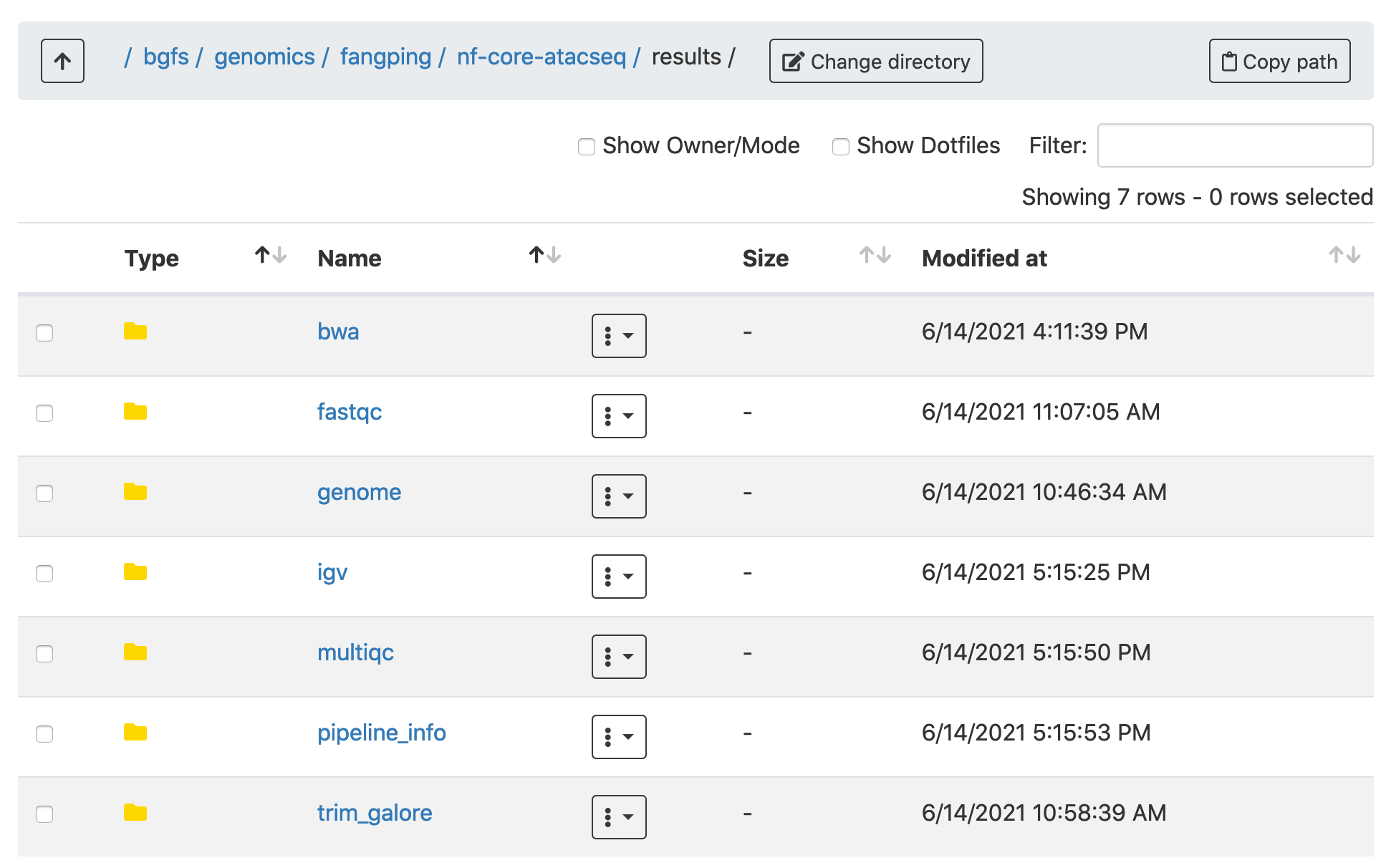
Navigate to /bgfs/genomics/fangping/nf-core-atacseq/results/multiqc/narrowPeak to go through the multiqc report.
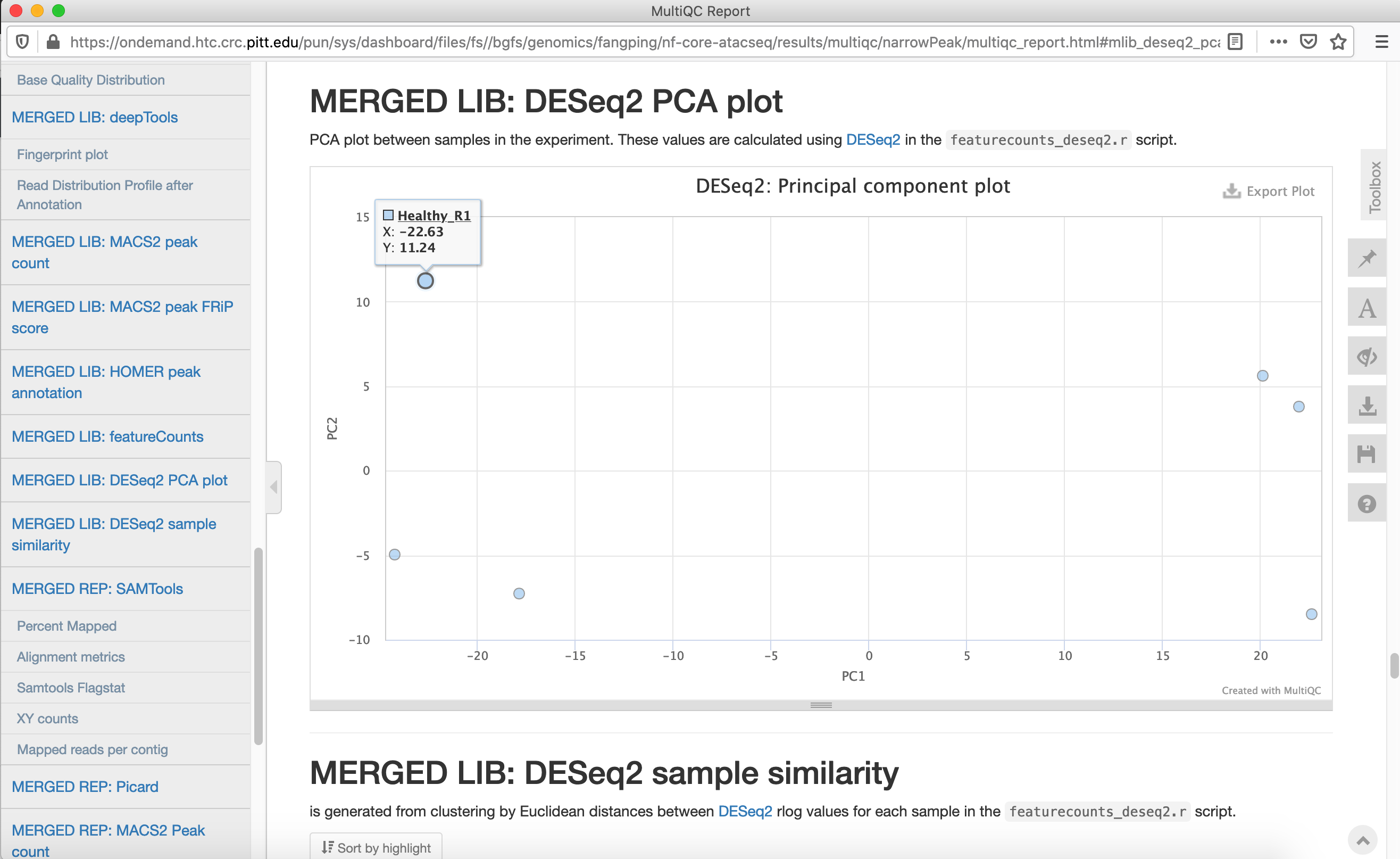
Click multiqc_report.html to open the report.
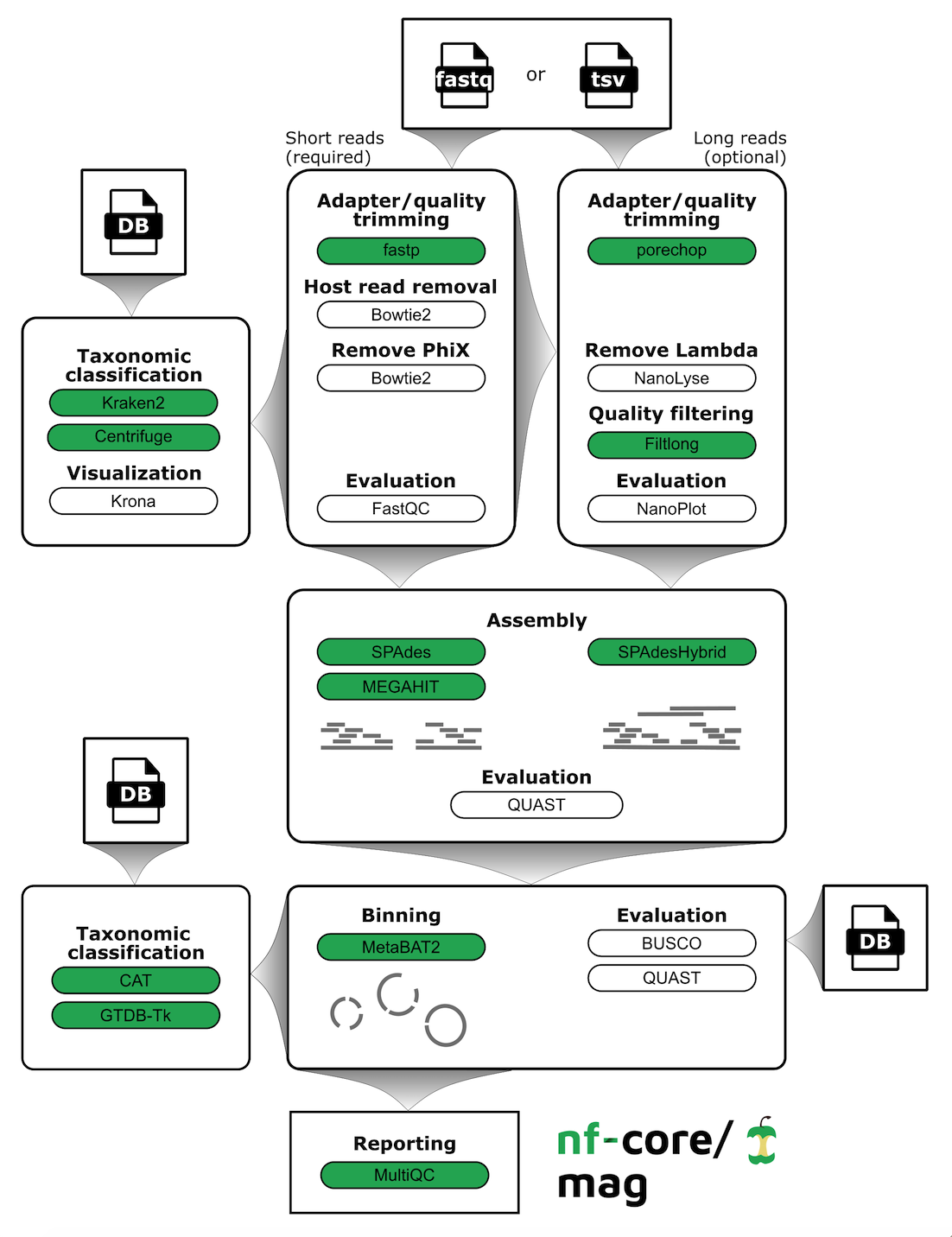
nf-core/mag
Assembly and binning of metagenomes. nf-core/mag is a bioinformatics best-practise analysis pipeline for assembly, binning, and annotation of metagenomes.
By default, the pipeline currently performs the following: it supports both short and long reads, quality trims the reads and adapters with fastp and Porechop, and performs basic QC with FastQC. The pipeline then:
- assigns taxonomy to reads using Centrifuge and/or Kraken2
- performs assembly using MEGAHIT and SPAdes, and checks their quality using Quast
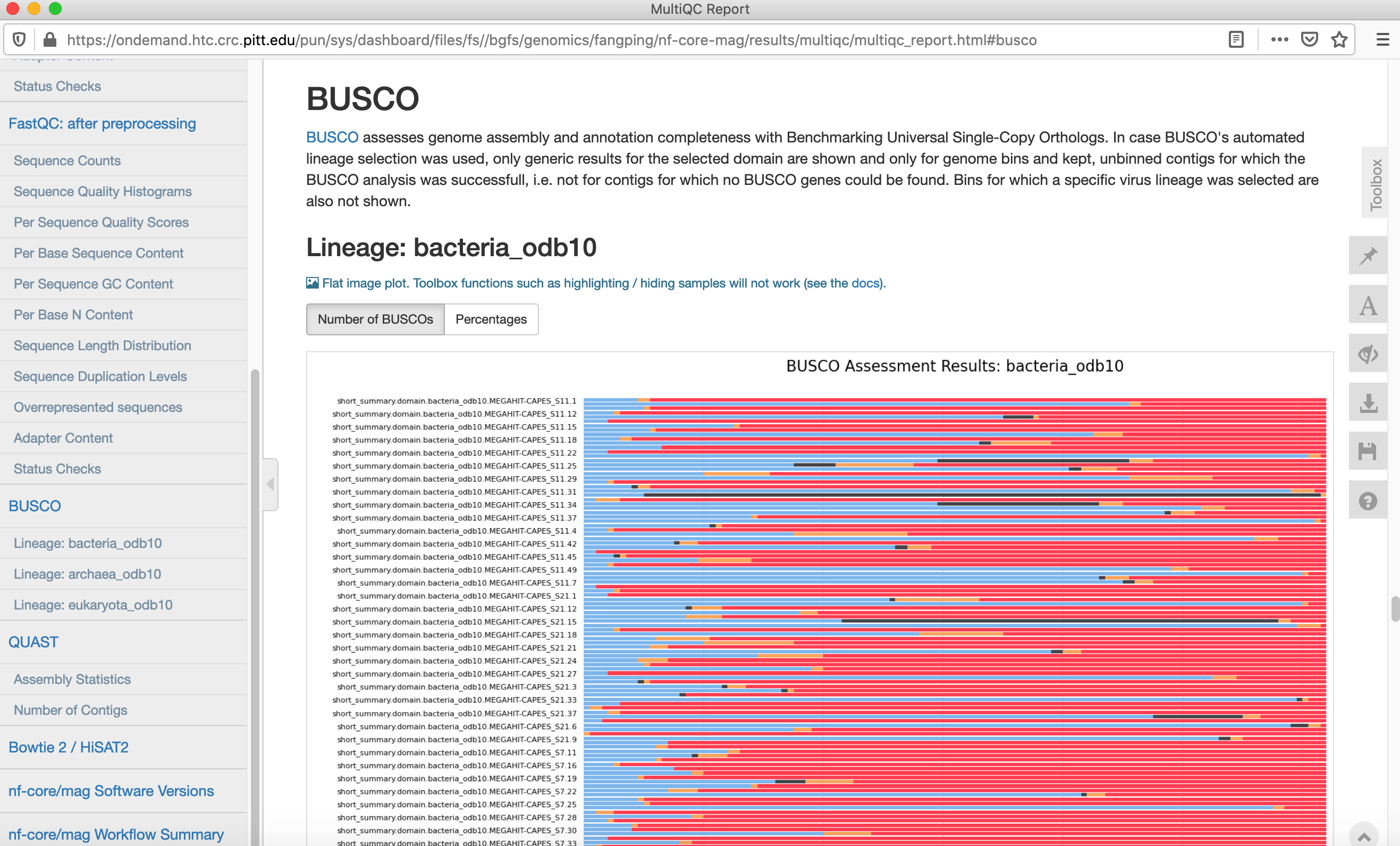
- performs metagenome binning using MetaBAT2, and checks the quality of the genome bins using Busco
- assigns taxonomy to bins using GTDB-Tk and/or CAT
Furthermore, the pipeline creates various reports in the results directory specified, including a MultiQC report summarizing some of the findings and software versions.
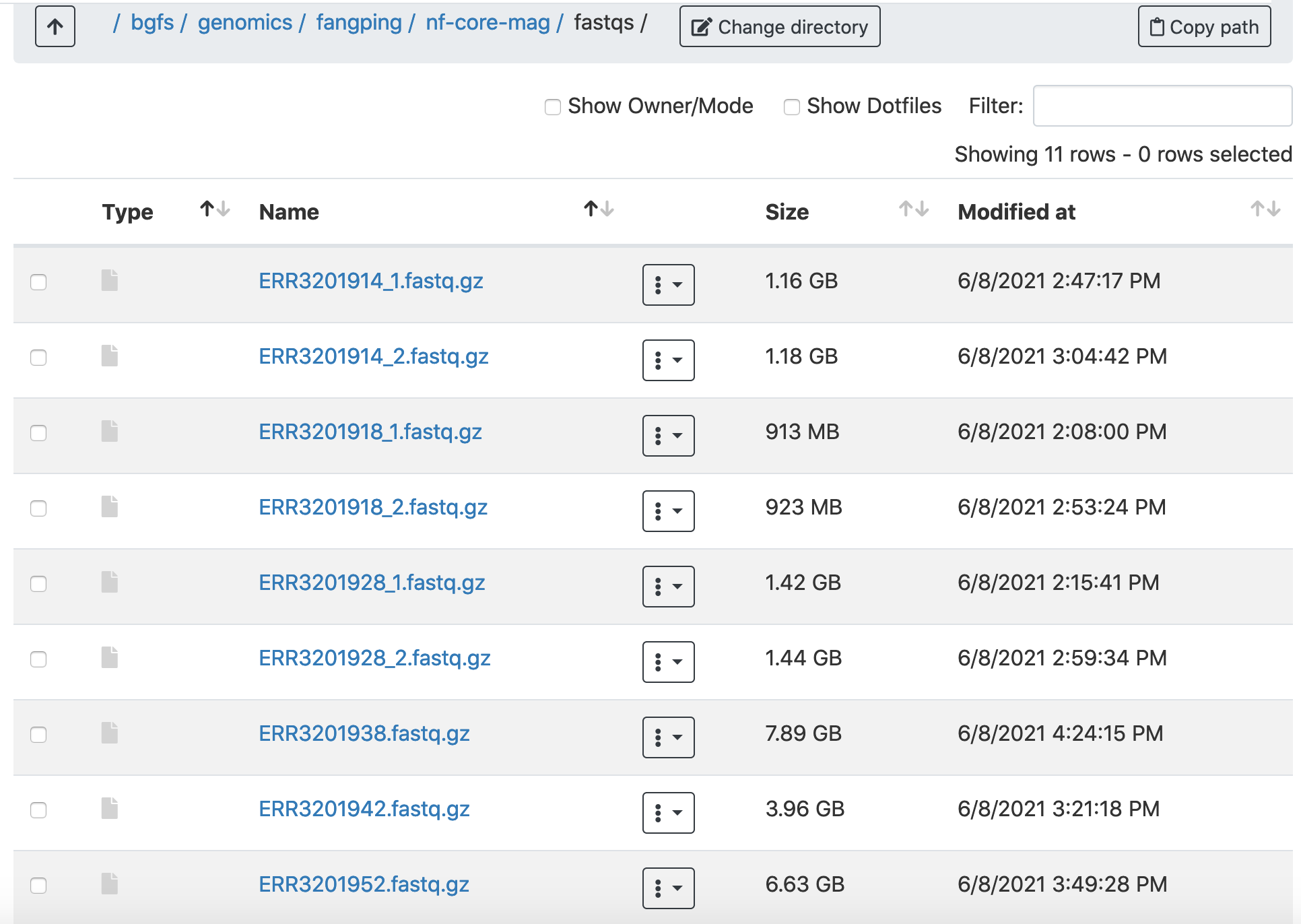
1. Logon ondemand.htc.crc.pitt.edu, Click Files -> Home Directory, Click Change directory and go to your bgfs or zfs folder. Test data for mag is in the folder /bgfs/genomics/fangping/nf-core-mag
2. Within the fastqs folder, I have downloaded the gut metagenome data of antibiotic-treated patients originating from Bertrand et al. Nature Biotechnology (2019). Three samples have been used. Note that sequencing data for the 3 samples have both Illumina and ONT reads.
| SAMPLE | ILLUMINA READS: ENA ID | ONT READs: ENA ID |
| CAPES_S7 | ERR3201914 | ERR3201938 |
| CAPES_S11 | ERR3201918 | ERR3201942 |
| CAPES_S21 | ERR3201928 |
ERR3201952 |
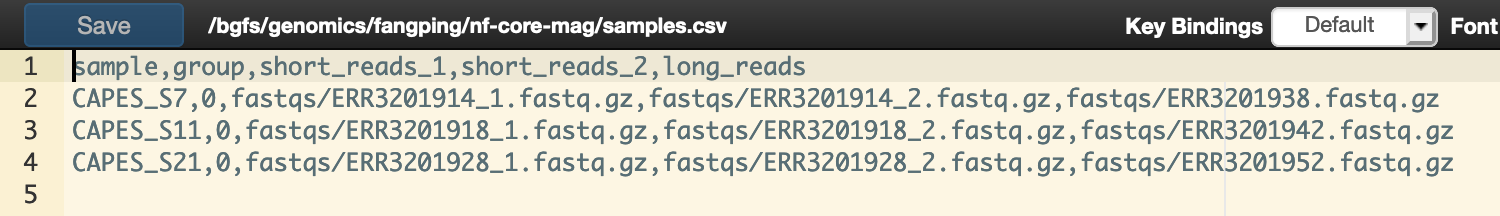
3.Samplesheet input file
To assign different groups or to include long reads for hybrid assembly with metaSPAdes, you can specify a CSV samplesheet input file that contains the paths to your FASTQ files and additional metadata.
This CSV file should contain the following columns:
sample,group,short_reads_1,short_reads_2,long_reads
The path to long_reads and short_reads_2 is optional. Example samplesheet is at /bgfs/genomics/fangping/nf-core-mag/samples.csv
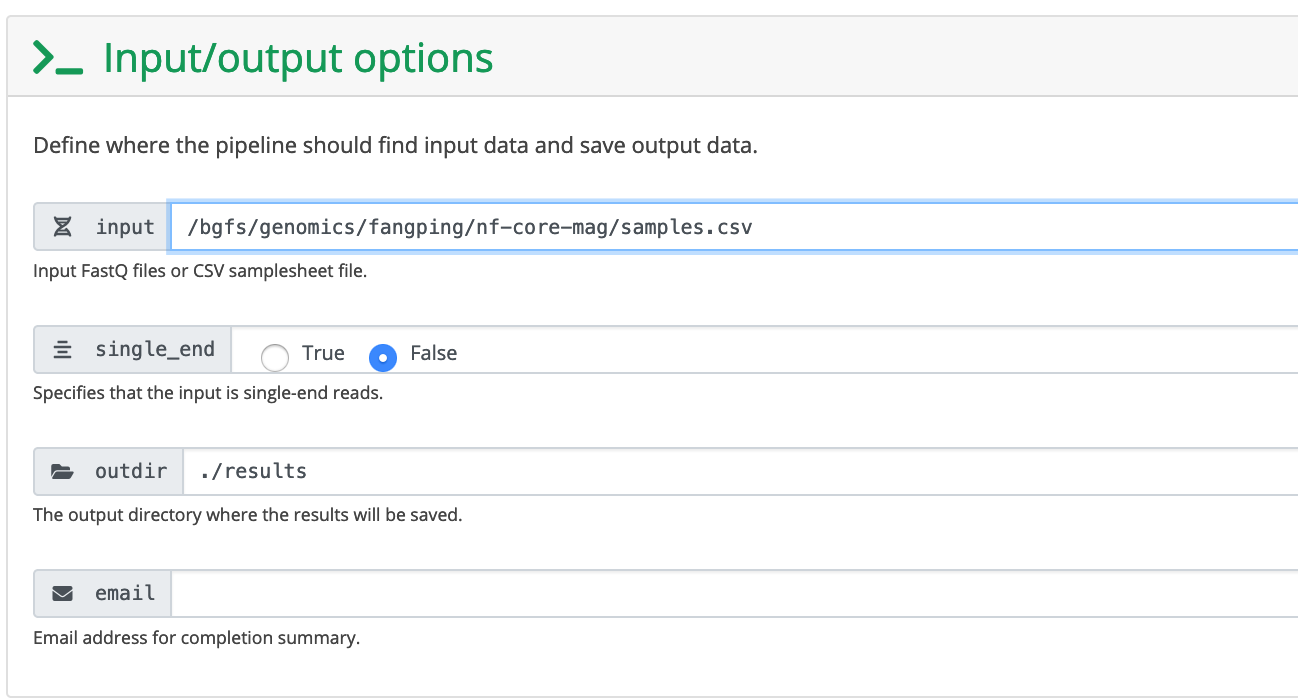
4. Fill the launch form
Click Genomics Apps -> nf-core pipelines, choose mag 2.0.0, click Launch, then click Connect to Nextflow
The input field is the absolute path to your sample sheet.
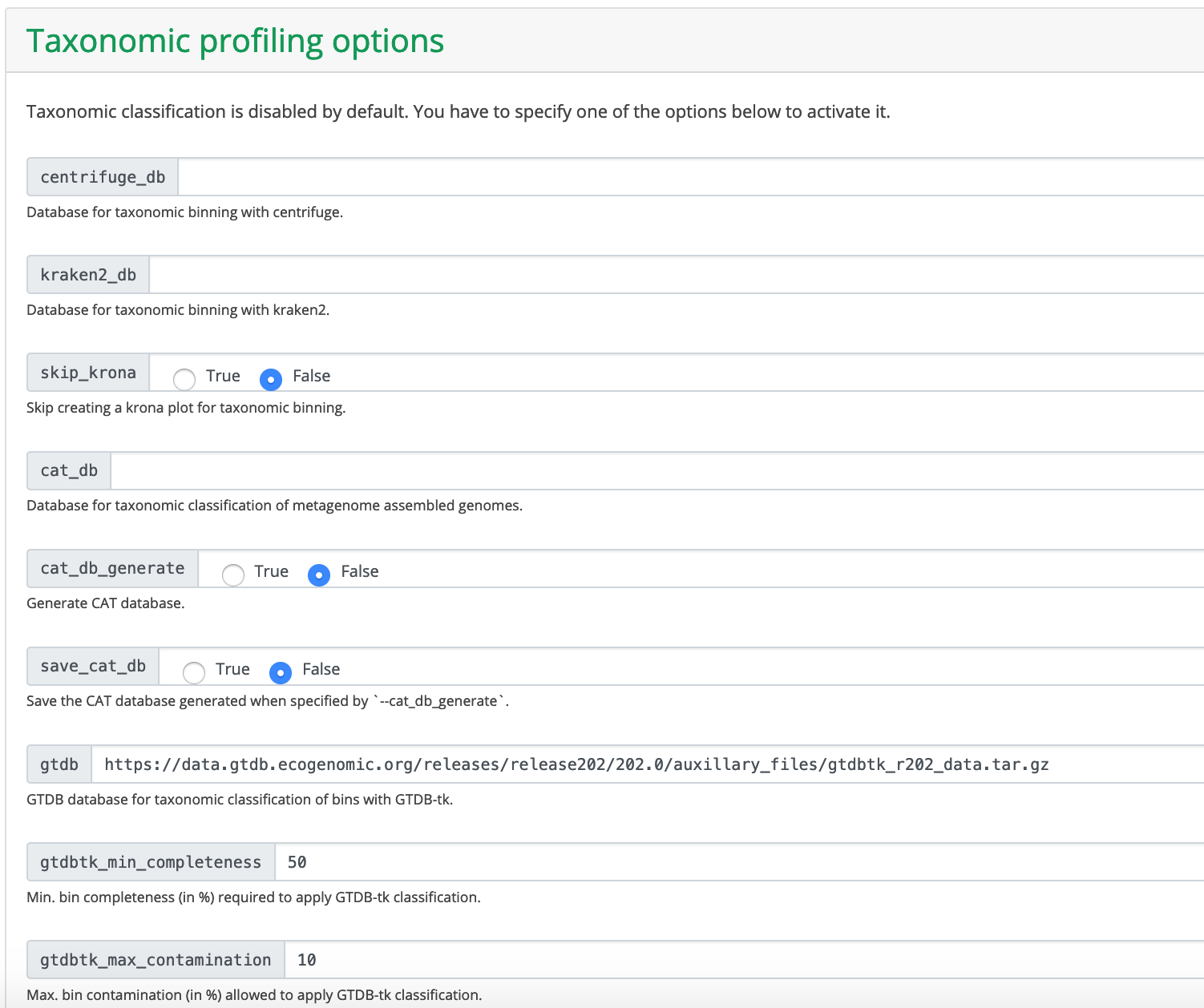
I have kept all other options as default values. Note that Taxonomic classification is disabled by default. cat_db, centrifuge_db and kraken2_db are available under /bgfs/genomics/refs/metagenome_refs
5. Lauch workflow
When you are ready, click Launch workflow.
6. Outputs
This document describes the output produced by the pipeline. Navigate to /bgfs/genomics/fangping/nf-core-mag/results to read the results.
Navigate to /bgfs/genomics/fangping/nf-core-mag/results/multiqc to view the multiqc results.
Click multiqc_report.html to read the multiqc report.